About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 54(5); 2011
Congenital heart disease in the newborn requiring early intervention
- Corresponding author: Sin Weon Yun, M.D. Department of Pediatrics, Chung-Ang University, College of Medicine, 224-1, Heukseok-dong, Dongjak-gu, Seoul 156-755, Korea. Tel: +82-2-6299-1478, Fax: +82-2-6263-2167, yswmd@cau.ac.kr
- Received April 06, 2011 Accepted April 25, 2011
- Abstract
-
Although antenatal diagnostic technique has considerably improved, precise detection and proper management of the neonate with congenital heart disease (CHD) is always a great concern to pediatricians. Congenital cardiac malformations vary from benign to serious conditions such as complete transposition of the great arteries (TGA), critical pulmonary and aortic valvular stenosis/atresia, hypoplastic left heart syndrome (HLHS), obstructed total anomalous pulmonary venous return (TAPVR), which the baby needs immediate diagnosis and management for survival. Unfortunately, these life threatening heart diseases may not have obvious evidence early after birth, most of the clinical and physical findings are nonspecific and vague, which makes the diagnosis difficult. High index of suspicion and astute acumen are essential to decision making. When patent ductus arteriosus (PDA) is opened widely, many serious malformations may not be noticed easily in the early life, but would progress as severe acidosis/shock/cyanosis or even death as PDA constricts after few hours to days. Ductus dependent congenital cardiac lesions can be divided into the ductus dependent systemic or pulmonary disease, but physiologically quite different from each other and treatment strategy has to be tailored to the clinical status and cardiac malformations. Inevitably early presentation is often regarded as a medical emergency. Differential diagnosis with inborn error metabolic disorders, neonatal sepsis, persistent pulmonary hypertension of the newborn (PPHN) and other pulmonary conditions are necessary. Urgent identification of the newborn at such high risk requires timely referral to a pediatric cardiologist, and timely intervention is the key in reducing mortality and morbidity. This following review deals with the clinical presentations, investigative modalities and approach to management of congenital cardiac malformations presenting in the early life.
- Introduction
- Introduction
Even though there are efforts to detect critical CHD in the fetal life or immediately after birth, large population of a neonate with heart defects remains undiagnosed until after developing serious manifestations.Because infant with such life-threatening heart defects may not initially have symptoms or the clinical sign may be obscure, serious condition may not be recognized on the routine physical examination in majority of cases1). Birth is a great event from fetal to the postnatal circulation; the most important changes are from an aquatic amniotic environment and placental gas exchange to breathing and pulmonary ventilation. Air breathing means sudden drop of pulmonary vascular resistance and marked increase in pulmonary blood flow. Fetal structures such as foramen ovale, ductus venosus and ductus arteriosus, which was vital for fetal circulation is no longer needed for survival and begin to close. Neonate with CHD associated with ductus-dependent pulmonary or systemic blood flow or with mixing physiology such as TGA is at a great risk of compromise and collapse as they fail to make an adequate transition2,3).
- Incidence
- Incidence
The incidence of CHD has been estimated to be 6-8/1,000 live births in the general population1-3). Infant's death from CHD accounts for around 3%4). Not all CHD may be detected before or at death. Critical cardiac defect with a high early mortality if not treated immediate after birth includes HLHS, coarctation of the aorta (COA) / interrupted aortic arch (IAA), TGA, TAPVR, critical aortic stenosis (AS), pulmonary atresia (PA) and tricuspid atresia (TA). Although individually rare, overall, CHDs contribute significantly to death in neonates. With early intervention, neonatal mortality from the heart can fall from 2-3/1000 to 0.6-0.8/1000 live births2,4). However, other associated important factors such as combined congenital anomalies, low birth weight, prematurity, lung problems, persistent pulmonary hypertensions, sepsis are also influenced in the overall outcome of newborn with CHD.
- Classification of CHD
- Classification of CHD
- 1. Life-threatening CHD
- 1. Life-threatening CHD
Structural cardiac malformations in which cardiovascular collapse is likely and compromised if not treated early. They include TGA, COA/IAA, AS, and HLHS/mitral atresia, PA and obstructed TAPVR.- 2. Clinically significant CHD
- 2. Clinically significant CHD
Structural cardiac malformations that have effects on heart function but where the collapse is unlikely to be need early intervention. Most common defects in this group are ventricular septal defect (VSD), complete atrioventricular septal defect (AVSD), atrial septal defect (ASD) and tetralogy of Fallot (TOF) with good pulmonary artery anatomy.- 3. Clinically non-significant CHD
- 3. Clinically non-significant CHD
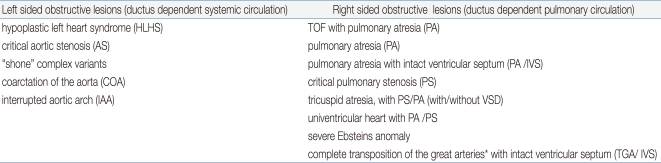
Anatomically defined cardiac malformations but no functional and clinical significance. They include small VSD, atrial septal defect (ASD), mild pulmonary stenosis (PS), only detectable with echocardiography and requiring no treatment.There are two types of the ductus dependent cardiac lesions. As listed the Table 1, ductus dependent systemic circulation (also called, left sided obstructed lesions) includes HLHS and its variants, severe AS, severe form of COA, IAA and its variants. These require ductal patency to maintain perfusion to the whole or even just the lower sides of the body, or the child develops progressive acidosis as the duct constricts. Consequently, perfusion falls and leg pulses become weak, impalpable and oliguria develop due to renal impairment and become progressively compromised5,9,10).The other type is the ductus dependent pulmonary circulation (also called, right sided obstructed lesions) which includes critical TOF, PA and its variants, critical PS, TA, with PS/PA (with/without VSD), univentricular heart with PS/PA, and severe form of Ebsteins anomaly7). TGA with intact ventricular septum (TGA/IVS) serve as ductus dependent lesion, but large ASD is more important to mixing of the circulation2,5). Most of these CHDs present progressive cyanosis without response in proper oxygen supply. Because their fetal physiology is chronically adapted to the hypoxia in the uterine life, newborn infants are able to tolerate some degree of cyanosis than older infants or children4). The variety of CHD is immense, because of lots of combinations of defects, which can affect the various cardiac levels, atrium, ventricle, septum, veins or great arteries10).Category of cyanotic CHD can be divided into decreased pulmonary flow with right to left shunting lesions (PA, TA with shunting at the atrial or ventricular level); poor mixing lesions (transposition physiology); and right to left shunt with intra cardiac mixing lesions (TAPVR, single ventriclular physiology, truncus arteriosus)4,7,10).Some CHD evolves during the fetal life as growth of cardiac structures is flow dependent. Thus, fetuses with mild left sided obstructive lesions may progress into coarctation/HLHS over time; similarly, pulmonary atresia with intact ventricular septum is considered a late phenomenon starting off as severe pulmonary stenosis. PPHN is another serious condition that is associated with other neonatal high risk factors5) which may be difficult to differentiate from the above mentioned cyanotic heart disease.
CHD can be classified into three main categories in clinical point of view4).
- Physiological change of the heart after birth
- Physiological change of the heart after birth
In the fetal life, oxygen and nutritional transport are provided by the placenta, which receives almost about 40% of the combined fetal cardiac output2). This returns to the fetal body via the umbilical vein and ductus venosus to the right atrium. The lungs have no function to oxygenation and receive only about 7% of fetal cardiac output. The remain of the right ventricular output being diverted via the ductus arteriosus to the descending aorta. That is, in a direction 'right to left shunt'. In addition, blood flows 'right to left' through the foramen ovale in the atrial level4). These two main communications allow to interrelate both sides of the heart, and it lets the wide variety of cardiac anomalies which turn out be critically serious in the postnatal life, able to sustain. However, after birth, the biggest change is the placenta not being in charge of circulation and both lungs being responsible for oxygenation route with the first few breaths.With the abrupt discontinuation from umbilical venous flow and the marked increase in return from lungs to the left atrium, the flap-like oval fossa virtually closes, prostaglandin-dependent ductus arteriosus also closes into ligamentum arteriosum, right and left hearts remain functionally independent. Ductal closure may be delayed if the baby has persistently significant cyanosis and acidosis3).
- Clinical manifestations
- Clinical manifestations
- 1. Dyspnea
- 1. Dyspnea
Persistent tachypnea or dyspnea may indicate lung or heart problems. Large shunt lesions manifest as dyspnea, tachypnea, feeding difficulty, irritability and distress. Ventilator weaning can be difficult in premature infants with large left to right cardiac shunts. Cyanosis with markedly reduced pulmonary blood flow usually leads to "quiet tachypnea", without significant respiratory distress1,2,5).- 2. Sign of poor perfusion
- 2. Sign of poor perfusion
Newborns with ductus dependent systemic circulatory lesions as mentioned above, develop progressive dyspnea, cold, clammy mottled skin, which indicates poor perfusion and acidosis, shock, oliguria due to end organ impairment and eventually progressive cardiovascular collapse at the time of ductal closure2,5,9). S3 gallop with or without significant cardiac murmur and cardiomegaly on chest x-ray may support these findings.- 3. Cyanosis
- 3. Cyanosis
In the neonate, decreased oxygen saturation is more often due to pulmonary than cardiac problems.Persistent hypoxia refractory to 100% oxygen supply would indicate cyanotic CHD rather than pulmonary problems. A hyperoxia test5) can be easily performed, using arterial blood gas analysis while 100% oxygen is supplied at a bed side. Increase in PO2 more than 220 mm Hg would suggest respiratory disease; 100-220 mm Hg would require evaluation for cyanotic CHD; less than 100 mm Hg would suggest cyanotic CHD and marked cyanosis less than 40-50 mm Hg would be likely to have a poor mixing disease such as TGA5). If hyperoxia test reveals positive or borderline results, immediate further evaluation for cyanotic CHD by the pediatric cardiologist is needed.Hyperoxia test may not always be helpful to differentiate coexisting heart and lung problems or PPHN with right to left shunt at both levels of atrium and ductus arteriosus.
The clinical sign of the critical heart defects in the neonate may be vague. Usually cardiac murmur is not helpful in this situation. It is very important for pediatricians to identify the newborn baby "not doing well" and having a high index of suspicion, and identifying the need for rapid cardiac evaluation and rule out a serious congenital cardiac problem which needs early intervention. They include persistent central cyanosis, unexplained acidosis, tachypnea without lung problems, etc5). Initial evaluation would include assessment of saturation monitoring, status of perfusion (blood gas analysis) and blood pressures in all extremities.The Table 1 lists various ductus dependent congenital cardiac malformations, which would require urgent prostaglandin infusion5). Some neonate with critical congenital cardiac lesions may present clinical symptoms first few hours to days of life with PDA closure. At this time, the babies would manifest severe acidosis/cyanosis/shock or even sudden death6).
- Diagnosis
- Diagnosis
- 1. Chest x ray
- 1. Chest x ray
When the neonate is suspected to have heart disease, chest roentgenogram is usually performed to rule out pulmonary disease as well as to evaluate pulmonary vascular marking and cardiomegaly. Some CHD has characteristic features such as "boot shaped heart" which could be seen in the TOF/PA and its variants2). This peculiar cardiac contour originates from concave small pulmonary trunk compared to the large aorta and right ventricular hypertrophy. Another specific feature called the "egg on string" appearance can be seen in TGA2,5). Narrow cardiac pedicle due to the almost anterior-posterior relationship of the transposed great vessels and "radiologic-absence of the thymus" make such peculiar cardiac border2,4). Pulmonary vascular markings depend on the degree of pulmonary stenosis and amount of pulmonary blood flow. Pulmonary venous congestion may suggest poor mixing at the atrial level with increased pulmonary blood flow due to a wide open ductus arteriosus. The characteristic "figure of 8" or "snowman appearance" in the supracardiac TAPVR could not be seen in the neonatal period and is rarely seen in the modern era of rapid diagnosis and early repair2,5). Instead of these classical features, most of the serious CHD which needs early intervention have no specific findings except vague cardiomegaly, change of pulmonary vascular marking and subtle finding of pulmonary venous congestion5-7).- 2. Electrocardiography (EKG)
- 2. Electrocardiography (EKG)
EKG has been considered a useful tool in the diagnosis of CHD, especially if echocardiogram is not easily available. An extreme left axis deviation (superior axis deviation) would point to AVSD in the acyanotic neonate and left axis deviation with left ventricular hypertrophy in the cyanotic baby would point to tricuspid atresia. Most of TOF and its variants manifest as right axis deviation (RAD) with right ventricular hypertrophy (RVH)2,5,7). However, since echocardiogram is easily available, the significance of EKG is decreasing. However, it is an invaluable diagnostic modality in the field of arrhythmias.- 3. Echocardiography
- 3. Echocardiography
Echocardiogram is the most valuable method in the diagnosis of CHD. More detailed identification of cardiac anatomy can be possible through two dimensional multiple views (including subcostal long and short axis, apical four chambers, parasternal long and short axis and suprasternal) which delineates the entire detailed anatomy in the various sections. Assessment of systolic ventricular function, measurement of chamber dimensions and wall thickness can be possible by M-mode echocardiography. Pulsed or continuous wave Doppler techniques can be used to assess the pressure gradients across the stenotic or regurgitation flow through the valves. Various Doppler wave forms can assess abnormal cardiac physiology; decreased flow in the descending aorta as seen in the COA; and estimation of pulmonary arterial pressure by measurement of the tricuspid regurgitation gradient. Color flow is a great tool in defining the direction of flow when valve regurgitation and shunt exist, the accentuation of flow across defects or narrowed valves, and it also helps detect abnormal turbulent flow such as coronary arteriovenous fistulas and collateral vessels5-7).- 4. Cardiac Catheterization
- 4. Cardiac Catheterization
Because of improved imaging technology, the diagnostic frequency of cardiac catheterization is relatively decreasing especially in the neonate. However, it is still the key in defining certain anatomic variants difficult to be delineated by echocardiography alone, such as RV to coronary arterial fistulae in PA/IVS, or aorto-pulmonary collateral arteries in the PA/VSD. Some cardiac surgeons prefer to confirm coronary anatomy by angiography in complete TGA prior to arterial switch operation.Therapeutic catheterizations are considered as one of the life saving modalities in some fields.
- Specific CHDs
- Specific CHDs
- 1. Aortic valve stenosis (AS)
- 1. Aortic valve stenosis (AS)
Aortic stenosis represents about 6% of CHD4). Valvular AS is described as restriction of blood flow through the aortic valve. It may present throughout life, but if presenting in the early newborn period it is physiologically and morphologically at the severe end of the spectrum. Stenotic aortic valve looks small and dysplastic and often bicuspid. Left ventricle may be markedly dilated with poor contraction or be hypertrophied when the systolic function is preserved. In some cases, left ventricle is poorly developed, similar to the borderline spectrum of HLHS. Clinical symptoms depend entirely on the severity of the obstruction and associated abnormalities such as subaortic stenosis or COA. Severity of the disease is correlated with the earlier presentation. Management also depends on the severity. Severe heart failure or other symptoms all require palliation by valvotomy through surgery or transcatheter methods. Early recognition before deterioration will inevitably lead to an improved outcome.- 2. Coarctation of the aorta (COA)
- 2. Coarctation of the aorta (COA)
COA represents 8% to 10% of CHD4). Major malformation is narrowing of distal part of the aortic arch commonly near the ductus arteriosus, and it is often accompanied by hypoplasia or diffuse narrowing of the aortic arch. In this case usually the lower half of the body is a ductus dependent and the symptoms will develop as it starts to close. About 40% of infants have other cardiac malformations and most of them present during the newborn period4). The most commonly associated malformation is VSD or bicuspid aortic valve, AS, other left sided obstructive lesions and more complex anomalies are also common. Severe COA may present with acidosis, congestive heart failure, renal impairment, cardiovascular collapse or even death. If the "differential saturation" (feet saturations being significantly lower than the arms) is present, this highly suggests that legs are ductus dependent, which includes critical COA/IAA, where the poorly saturated pulmonary arterial blood flow to the lower part of the body via the PDA. In this situation, differential diagnosis is needed with PPHN. If there is difference of blood pressure between arms and legs (right arm pressure being significantly higher than the feet), arch obstructive lesions are highly suggestive. If the baby is in a significant shock, pressures can be weakened in all extremities4,10). If ductal dependent clinical presentations are detected early, infants can survive with prostaglandin E1 (PGE1) treatment.Definitive early surgery is preferred. Operative mortality of simple COA repair is low but overall outcome depends on associated cardiac malformations.- 3. Hypoplastic left heart syndrome (HLHS)
- 3. Hypoplastic left heart syndrome (HLHS)
HLHS is rare, accounting only for 2-3% of all CHD11). By definition, the left side of the heart is unable to support systemic circulation.HLHS includes aortic valve atresia and some forms of mitral atresia4). Absent forward flow through the left ventricular outlet is characteristic. Left ventricle is markedly underdeveloped and often rudiment. Aortic arch is also hypoplastic and ascending aorta is very small, acting simply as a passage into the coronary arteries. Pulmonary venous return can only reach systemic circulation by traversing the patent foramen ovale to reach the right atrium. This implies mixing of pulmonary venous and systemic venous flow, creating a mild cyanotic condition11).All systemic circulation is absolutely dependent to the ductus arteriosus. After birth, systemic vascular resistance is higher than pulmonary, with the ductal closure after birth, nonfunctioning left ventricle cannot take charge of the cardiac output. This leads to circulatory deterioration, metabolic acidosis, and shock. Increased pulmonary flow leads to increase in the left atrial pressure and subsequent pulmonary edema5,10).Clinical presentations are early heart failure, listlessness, duskiness, tachypnea or even death due to circulatory collapse. The median age at diagnosis is about two days of life. The natural history is early death with almost no prospect of prolonged natural survival. PGE1 infusion is necessary to survive. In previous years HLHS showed high mortality because of the poor interventional outcome. However, in recent years, radical palliative surgery (Norwood and its variants) has become more widespread and the outcome has improved10,11).- 4. Interruption of the aortic arch (IAA)
- 4. Interruption of the aortic arch (IAA)
In IAA, failure of development in a portion of the aortic arch is the major pathology, so there is no direct connection between ascending and descending aorta. Lower half of the body has an entirely ductus-dependent circulation. The interruption may be distal to the left subclavian artery (type A) or between the left common carotid and the left subclavian artery (type B) or between the innominate artery and the left carotid artery (Type C)4). IAA is always associated with other anomalies such as VSD, truncus arteriosus, aortopulmonary window, or other complex anomalies. Type B is commonly associated with 22q11 deletion (DiGeorge syndrome).Because of associated other cardiac anomalies, one-third of babies present before the routine examination. Infants with IAA deteriorate very quickly once their duct begins to close, with initial breathlessness, congestive heart failure, acidosis, cardiovascular collapse, and death within few days.Early recognition and intervention will inevitably lead to a better outcome. Surgery is primary repair of the aortic arch and what else is done depends on associated other anomalies.- 5. Pulmonary atresia (PA)
- 5. Pulmonary atresia (PA)
Main pathology in PA is the absence of a direct connection between the right ventricle and the lungs. Two main types are pulmonary atresia with ventricular septal defect (PA/VSD), pulmonary atresia with intact ventricular septum (PA/IVS)4, 6).In PA/VSD, also known as tetralogy of Fallot type pulmonary atresia, there are usually favorable sized two ventricles with large subaortic VSD and variable source of pulmonary arterial supply.Some patients have only ductal structure, but more commonly have major aorto pulmonary collateral arteries (MAPCAs) arising from the descending aorta. PA may also be a spectrum of more complex cardiac malformations such as, congenitally corrected TGA or heterotaxy syndrome or single ventricle2,5,6).In the PA/IVS, tricuspid valve and right ventricle are usually severely underdeveloped, but pulmonary arteries are relatively well developed and supplied by PDA5,7). Infants with PA/IVS- a typical form of 'ductus-dependent pulmonary circulation' - become more cyanosed and aggravated as the ductal closes, and if without prostaglandin infusion, eventually will collapse and perhaps die within a first few day of life. PA/IVS has a category of functionally single ventriclular physiology, and the eventual surgical goal may usually be 'Fontan' type operation, more often a cavopulmonary shunt achieving a right heart bypass. Early palliation involves shunt such as Blalock-Taussig (BT) shunt to replace the ductus.Because less ductal dependent than PA/IVS, the natural course of PA/VSD depends on the pulmonary blood supply and other variables. Affected infant may present cyanosis in the early life, but some can survive into adult life, even without any intervention if the various pulmonary supplies are appropriate. However other problems develop in later life, leading to poor quality of life and shortened life span4-6). Management of PA/VSD depends on the pulmonary blood supply. About half of those will be suitable for corrective surgery with VSD closure and connect the right ventricle to the pulmonary arteries using the conduit placement2,4,5,7).- 6. Total anomalous pulmonary venous return (TAPVR)
- 6. Total anomalous pulmonary venous return (TAPVR)
TAPVR represents around 1% of CHD2,15). All four pulmonary venous directly connect to the right atrium instead of the left atrium. There are four main types according to the connection2,4). 1) supracardiac type (50%) to the innominate vein, 2) infradiaphragmatic type (20%) to the hepatic or portal vein, 3) cardiac type (20%) to the coronary sinus, 4) mixed type (10%) combination of any of type2,4,15). The timing and mode of presentation depend on the type and degree of obstruction. Infracardiac type is the most commonly obstructed and may have serious manifestations. Survival rate is reported 50% at 1-month and 0% at 12-month without treatment15).Common pulmonary venous channels are delivered to the right atrium, and there the mixing of the pulmonary and systemic circulations occurs. Systemic desaturation occurs as the result of mixing of two circulations. In patients with TAPVR with obstruction, progressive cyanosis and respiratory distress dominate the presentation. No positive physical findings except loud and single S2 and/or gallop sound is noted. EKG finding is RAD and RVH. Infants with obstructed TAPVR often have unremarkable clinical findings except progressive cyanosis and dyspnea, they frequently masquerade as parenchymal lung disease with pulmonary hypertension such as congenital pneumonia, meconium aspiration pneumonitis, respiratory distress syndrome and pulmonary lymphangiectasia. Unless definite evidence for parenchyme lung lesions is present, or there is no appropriate response to conventional therapeutic measures, early echocardiography should be considered to make these distinctions10,15).Infants may not present immediately after birth, but most affected babies present with the combination of cyanosis, heart failure and deterioration by the time the confirmative diagnosis is made when obstructed. The only option is early primary repair as soon as possible. Prostaglandin infusion is not helpful in this case, even harmful. It leads to increases pulmonary blood flow and reduces the pulmonary vascular resistance and may exacerbate the pulmonary venous congestion when obstruction is combined2,10).Long-term surgical and overall survival outcome is fairly good although in some cases, there is recurrent and fatal pulmonary venous stenosis. Perioperative mortality is related with the clinical conditions, although it is improved in recent years, but it is still significant.For these reasons, early diagnosis can potentially improve an overall outcome by ensuring better preoperative status.- 7. Transposition of the great arteries with intact ventricular septum (TGA/IVS)
- 7. Transposition of the great arteries with intact ventricular septum (TGA/IVS)
TGA represents around 5-8% of CHD and is one of the most common cyanotic heart diseases in the early presentation2,16). There are many variations of the disease, but cardiac malformation is characterized by atrioventricular concordance and ventriculoarterial discordance (the aorta arises from the right ventricle, pulmonary trunk arises from the left ventricle)16). In 50% of the cases, the TGA is an isolated finding. This condition is designated as 'simple' or 'complete' TGA or TGA/IVS. By contrast, complex transposition includes all the cases with coexisting malformations, such as VSD, PS, left ventricular outflow tract obstruction, aortic arch anomalies, and anomalous venous systemic return. The pulmonary arterial oxygen contents are significantly higher than the aorta, so hyperoxemic blood traveling within the pulmonary circulation is inefficient and on the other hand, significant hypoxic blood goes through the systemic circulation. If the ventricular septum is intact or ASD is restrictive, limited intercirculatory mixing leads to progressive and profound cyanosis within the first few hours of life. Its severity and onset depend on the degree of mixing between the two circulations. Complete separation of two circulatory systems will lead to death. After birth some cross-flow between two separated circulations such as ductus or foramen ovale are necessary to survive. Without prompt treatment, most infants would die soon within a few days. There will be a loud, single S2, but no specific murmur is notable. The EKG will show RAD and RVH. On the chest X-ray, cardiomegaly with increased pulmonary vascular markings and so called" egg-shaped heart" with a narrow mediastinum is the characteristic findings on chest x-ray2), but not always2,16).Congestive heart failure with dyspnea in addition to intractable cyanosis which does not respond to the oxygen is common presentations. Acidosis as well as hypocalcemia and hypoglycemia are also frequent. If large VSD or PDA coexists, congestive heart failure with dyspnea and feeding difficulties in addition to the cyanosis is a common manifestation in the first week of life, and eventually progress into obstructive pulmonary vascular disease2). Babies with TGA should be started on PGE1 infusion to maintain ductal patency which increases the pulmonary flow, and leads to pulmonary venous return and left atrial pressure, thus promoting left to right shunt at the atrial level16,17). If the foramen ovale is restricted, PGE1 alone could not achieve clinical improvement and emergency balloon atrial septostomy (Rashkind balloon septostomy) is the only way to rescue these infants.
- Management
- Management
- 1. Ductus dependent pulmonary circulatory lesions
- 1. Ductus dependent pulmonary circulatory lesions
Right sided lesions such as TOF and PS/PA and its variants usually represent as central cyanosis. The degree of cyanosis would depend on the patency of the ductus. Hyperoxia test with arterial blood gas would help differentiating between respiratory and cardiac origin6). Cyanosis may aggravate with constriction of the ductal tissue. These infants require prompt start on PGE1 infusion to keep widely open the ductus arteriosus until Blalock Taussig (BT) shunt surgery, which helps to augment pulmonary flow for PA.Airway management is always a primary concern and PGE1 infusion is also the key in the management which decreases pulmonary vascular resistance and enhances the left to right shunting and eventually increases pulmonary blood flow2). The initial intravenous dose of PGE1 is 0.05 µg/kg/min. If no improvement, increment to 0.1 µg/kg/min. After the infant's condition has stabilized, the usual maintenance dose of PGE1 is 0.025 µg/kg/min. Apnea, bradycardia, hypotension, fluid-electrolyte imbalances, irritablilty, fever and cutaneous flushing are potential complicating side effects of PGE1 so management of the airway is essential along with determining if the patient is not possibly septic. Apnea secondary to prostaglandin is not a rare indication for tracheal intubation, but do not reduce the dose and never stop. Long-term use is associated with cortical hyperostosis, an effect that does not seem to be dose related. Monitoring in an intensive care unit is therefore, advised18). Although there is no CHD for which PGE1 is contraindicated, obstructed TAPVR and TGA with a restrictive atrial septum may exacerbate with PGE12,10).- 2. Ductus dependent systemic circulation lesions
- 2. Ductus dependent systemic circulation lesions
Left sided lesions such as HLHS, AS, COA and its variants depend on the ductus to maintain systemic perfusion. Sign of poor perfusion, diminished leg pulses, shock is common, and the clinical presentations can mimic that of sepsis.Oxygen supplementation can exacerbate the closure of the ductus arteriosus and worsen the infant's condition by cardiogenic shock. Therefore, do not increase oxygen supply until after PGE1 has been started. In this patients treatment should aim to optimize systemic oxygenation and prevent metabolic acidosis, which can be harmful to perioperative condition.Two fundamental principles underlie the management of these patients. Firstly, maintain ductal patency (to provide systemic perfusion). PGE1 infusion is vital for survival. Secondly, when ductal patency has been prepared, attention should be directed to the flow balance between the pulmonary and systemic circulation. Increased pulmonary blood flow lead to decrease in systemic and myocardial flow. Signs of poor perfusion are oliguria, metabolic acidosis and myocardial dysfunction. Perioperative management brings together the most challenging fields of neonatal cardiac intensive care with the aim of balancing systemic and pulmonary circulations. Ventilatorory strategies must aim to increase pulmonary vascular resistance to avoid pulmonary over circulation. Maintaining the balance between two competitive circulations allows adequate perfusion to the systemic and myocardium and the ideal pulmonary to systemic flow ratio are of about 1:1. This goal can be achieved by meticulous adjusting by a modest positive end expiratory pressure (PEEP) (4-6 cm H2O), modulate inspiratory rate, pressures or tidal volumes to keep an arterial CO2 tension of 5-6 kPa, avoid too much oxygen supply, maintain systemic arterial saturation around 80% and avoid respiratory alkalosis10,23). The risk of high FiO2 or low partial pressure of CO2 (paCO2) is that these might lower pulmonary vascular resistance, may lead to volume loading to the pulmonary circulation, and lead to cardiac failure11,12). Sedation with morphine is usually necessary; muscle relaxants should be considered for infants in shock who sustains tachypnea10).Recent focus has been given to lowering systemic vascular resistance to secure better systemic perfusion, using vasodilators such as phenoxybenzamine and the phosphodiesterase inhibitor, milrinone13). To optimize the oxygen delivery, mixed venous saturations should be monitored closely, and some investigators have advocated the use of continuous in-line monitoring of mixed venous saturations11,14).In spite of all these effort, if low cardiac output persists, adequacy of prostaglandin infusion, intravascular volume and presence of anemia should be reassessed. If the blood pressure is permissible, a low dose nitroprusside infusion can lead to improved metabolic acidosis. Otherwise, low dose infusion of inotrope may be benefit to cease the vicious cycle.Generally, high dose inotrope infusions should be avoided because it may increase systemic vascular resistance, which may aggravate the flow distribution balance10).Functionally univentricular palliative treatment consists of three stages: (1) Neonate: Norwood operation, (2) 6-8 months of age: stage II, (3) 18 months and 5 years of age: stage III.HLHS is still regard as one of the highest-mortality lesions. Although surgical outcomes continue to improve, survival is currently around 65% at 5 years of age and 55% at 10 years of age11).- 3. Transposition of the great arteries with intact ventricular septum (TGA/IVS)
- 3. Transposition of the great arteries with intact ventricular septum (TGA/IVS)
Balloon atrial septostomy (BAS), so called Rashkind balloon septostomy is the procedure that involves locating balloon-tipped catheter in the left atrium, via oval fossa. After inflating the balloon, it is pulled back into the right atrium, to tear the friable atrial septum, and if the final diameter of ASD is more than 5 mm, and flap motion of the inferior rim of the atrial septum increases and increase in the oxygen saturation is noted, the procedure is satisfactory18,19). This is an effective and safe procedure for creating long lasting adequate interatrial communications.In addition, other supportive measures such as ventilator settings and oxygen supply are necessary to optimize the clinical condition. Aggressive conservative care may lead to disturbance of intercirculatory shunts, which may worsen the baby. High oxygen supply may accelerate ductal closure, may lead to adverse effect to intercirculatory mixing and aggressively high ventilator mode (positive inspiratory pressure (PIP) and PEEP) can lead to excessive alveolar inflation, and therefore, it should be avoided16,24).Corrective surgery with arterial switch (Jatene) is the surgery of choice, which will achieve complete anatomical and physiological correction within few days of life. Its supremacy has been corroborated by long-term follow up that shows preservation of the left ventricular function, normal sinus rhythm and low mortality with a survival rate of 88% at both 10 and 15 years21,22).Early diagnosis and prompt management are the only way to save these patients.Therapeutic cardiac catheterization may be required to perform the emergency BAS in TGA/IVS, which can dramatically improve oxygen saturations by allowing mixing in the atrial level, and the infant can wait in a better clinical condition for the arterial switch operation. Transcatheter dilatation with the balloon (valvuloplasty) for critical PS or AS is technically difficult, but is performed as rescue procedures. In the severe COA, dilatation with balloon is often life saving, but rate of restenosis is very high, so the first line therapy should be surgical repair2,5-7).All the patients also require meticulous general supportive care to improve the overall outcomes. These include vascular access, airway management, inotropics, sepsis control, fluid, acid-base, electrolyte balance, temperature control, nutritional support and, etc.When prenatal diagnosis is made, planned delivery in a tertiary center is very important to improve overall clinical outcome and survival.
- Conclusion
- Conclusion
In this era with marked improvement of antenatal and postnatal diagnosis, accurate detection of symptomatic neonates with suspected CHD still remains difficult for pediatricians.Pediatricians must always have a high index of suspicion for congenital cardiac malformations in which early intervention is crucial. If the patient is in shock or shows cyanosis in spite of proper management, CHD should be suspected. Early detection and prompt intervention for these critically ill patients are the only way to save lives. Many of these babies need to be transferred to tertiary centers. Early discussion with the pediatric cardiology center is needed about the appropriate measure in suspected infants to avoid the pursuit of other less likely diagnoses. Management for such conditions may worsen the clinical situation of the patient before and during transfer. If the fetus has above mentioned CHD in the antenatal echocardiography, or serious postnatal outcomes are anticipated, planned delivery in a tertiary center capable of managing is encouraged.
- References
- 1. Chang RK, Gurvitz M, Rodriguez S. Missed diagnosis of critical congenital heart disease. Arch Pediatr Adolesc Med 2008;162:969–974.2. Yee L. Cardiac emergencies in the first year of life. Emerg Med Clin North Am 2007;25:981–1008.
[Article]3. Friedman AH, Fahey JT. The transition from fetal to neonatal circulation: normal responses and implications for infants with heart disease. Semin Perinatol 1993;17:106–121.4. Knowles R, Griebsch I, Dezateux C, Brown J, Bull C, Wren C. Newborn screening for congenital heart defects: a systematic review and cost-effectiveness analysis. Health Technol Assess 2005;9:1–152. iii–iv.5. Nadas AS, Fyler DC. In: Keane JF, Lock JE, Fyler DC,Hypoxemia. editors. Nada's pediatric cardiology. 20062nd edition. Philadelphia: Saunders Elsevier, :97–101.6. Krishnan US. Approach to congenital heart disease in the Neonate. Indian J Pediatr 2002;69:501–505.7. Gewitz MH, Woolf PK. In: Fleisher GR, Ludwig S,Cardiac emergencies. editors. Textbook of pediatric emergency medicine. 20065th edition. Philadelphia: Lippincott Williams and Wilkins, :717–718.8. Waldman JD, Wernly JA. Cyanotic congenital heart disease with decreased pulmonaryblood flow in children. Pediatr Clin North Am 1999;46:385–404.9. Fedderly RT. Left ventricular outflow obstruction. Pediatr Clin North Am 1999;46:369–374.10. Penny DJ, Shekerdemian LS. Management of the neonate with symptomatic congenital heart disease. Arch Dis Child Fetal Neonatal Ed 2001;84:F141–F145.11. Barron DJ, Kilby MD, Davies B, Wright JG, Jones TJ, Brawn WJ. Hypoplastic left heart syndrome. Lancet 2009;374:551–554.12. Hoffman GM, Tweddell JS, Ghanayem NS, Mussatto KA, Stuth EA, Jaquis RD, et al. Alteration of the critical arteriovenous oxygen saturation relationship by sustained afterload reduction after the Norwood procedure. J Thorac Cardiovasc Surg 2004;127:738–745.13. Tweddell JS, Hoffman GM, Fedderly RT, Berger S, Thomas JP Jr, Ghanayem NS, et al. Phenoxybenzamine improves systemic oxygen delivery after the Norwood procedure. Ann Thorac Surg 1999;67:161–167.14. Li J, Zhang G, McCrindle BW, Holtby H, Humpl T, Cai S, et al. Profiles of hemodynamics and oxygen transport derived by using continuous measured oxygen consumption after the Norwood procedure. J Thorac Cardiovasc Surg 2007;133:441–448.15. Seale AN, Uemura H, Webber SA, Partridge J, Roughton M, Ho SY, et al. Total anomalous pulmonary venous connection: morphology and outcome from an international population-based study. Circulation 2010;122:2718–2726.16. Martins P, Castela E. Transposition of the great arteries. Orphanet J Rare Dis 2008;3:2717. Mair DD, Ritter DG. Factors influencing intercirculatory mixing in patients with complete transposition of the great arteries. Am J Cardiol 1972;30:653–658.18. Lin AE, Di Sessa TG, Williams RG. Balloon and blade atrial septostomy facilitated by two-dimensional echocardiography. Am J Cardiol 1986;57:273–277.19. Perry LW, Ruckman RN, Galioto FM Jr, Shapiro SR, Potter BM, Scott LP. Echocardiographically assisted balloon atrial septostomy. Pediatrics 1982;70:403–408.20. Thanopoulos BD, Georgakopoulos D, Tsaousis GS, Simeunovic S. Percutaneous balloon dilatation of the atrial septum: immediate and midterm results. Heart 1996;76:502–506.21. Losay J, Touchot A, Serraf A, Litvinova A, Lambert V, Piot JD, Lacour-Gayet F, Capderou A, Planche C. Late outcome after arterial switch operation for transposition of the great arteries. Circulation 2001;104:I121–I126.22. Choi BS, Kwon BS, Kim GB, Bae EJ, Noh CI, Choi JY, Yun YS, Kim WH, Lee JR, Kim YJ. Long-term outcomes after an arterial switch operation for simple complete transposition of the great arteries. Korean Circ J 2010;40:23–30.23. Tálosi G, Katona M, Rácz K, Kertész E, Onozó B, Túri S. Prostaglandin E1 treatment in patent ductus arteriosus dependent congenital heart defects. J Perinat Med 2004;32:368–374.24. Atz AM, Feinstein JA, Jonas RA, Perry SB, Wessel DL. Preoperative management of pulmonary venous hypertension in hypoplastic left heart syndrome with restrictive atrial septal defect. Am J Cardiol 1999;83:1224–1228.