About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 54(11); 2011
Familial hyperkalemic periodic paralysis caused by a de novo mutation in the sodium channel gene SCN4A
- Corresponding author: June-Bum Kim, MD, PhD. Department of Pediatrics, Konyang University College of Medicine, 685 Gasuwon-dong, Seo-gu, Daejeon 302-718, Korea. Tel: +82-42-600-9230, Fax: +82-42-600-8835, hoppdoctor@hanmail.net
- Received April 05, 2011 Revised June 07, 2011 Accepted July 06, 2011
- Abstract
-
Familial hyperkalemic periodic paralysis (HYPP) is an autosomaldominant channelopathy characterized by transient and recurrent episodes of paralysis with concomitant hyperkalemia. Mutations in the skeletal muscle voltage-gated sodium channel gene SCN4A have been reported to be responsible for this disease. Here, we report the case of a 16-year-old girl with HYPP whose mutational analysis revealed a heterozygous c.2111C>T substitution in the SCN4A gene leading to a Thr704Met mutation in the protein sequence. The parents were clinically unaffected and did not have a mutation in the SCN4A gene. A de novo SCN4A mutation for familial HYPP has not previously been reported. The patient did not respond to acetazolamide, but showed a marked improvement in paralytic symptoms upon treatment with hydrochlorothiazide. The findings in this case indicate that a de novo mutation needs to be considered when an isolated family member is found to have a HYPP phenotype.
- Introduction
- Introduction
Familial hyperkalemic periodic paralysis (HYPP, online mendelian inheritance in man [OMIM] no. 170500) is an autosomal-dominant pathology of the sodium channels that results in reversible flaccid paralysis with episodic hyperkalemia1). The symptoms usually begin in the first decade of life, presenting as bouts of mild-to-severe muscle weakness lasting for minutes to hours. The paralytic attacks occur most frequently in the morning and are commonly triggered or aggravated by fasting, potassium-rich foods, emotional stress, or rest after vigorous exercise1,2). Mutations in the SCN4A gene (OMIM no. 603967), which encodes the alpha subunit of the skeletal muscle sodium channel, are responsible for the majority of HYPP cases3).To our knowledge, de novo mutations have not yet been reported in HYPP. Described here is a case of HYPP in a patient with an apparently sporadic presentation who carries a de novo mutation in the SCN4A gene. This mutation predicts a codon change from threonine to methionine at residue 704 (Thr704Met, c. 2111C>T). This is the first report describing a spontaneous mutation in the SCN4A gene in a patient with HYPP whose parents neither possessed nor transmitted the same mutation.
- Case report
- Case report
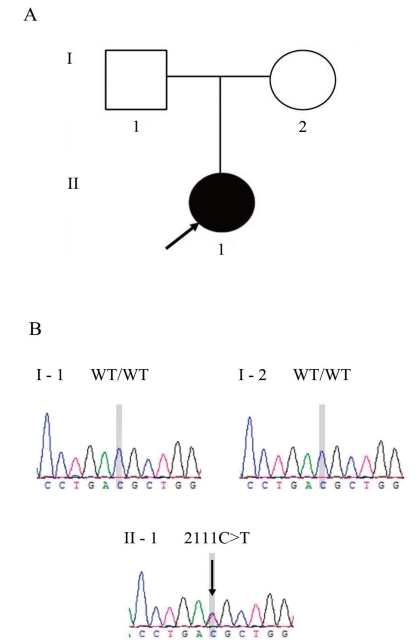
A 16-year-old girl was referred to our clinic for evaluation of periodic muscle weakness experienced over the past 7 years. During the early stages, her paralytic attacks occurred at a frequency of once a month, but recently, they occurred 2 or 3 times a week, and generally lasted from at least a few minutes up to 3 hours. The muscle weakness was often generalized. Acute crises often followed the ingestion of orange juice or watermelon, stressful circumstances, fatigue, and oversleeping. Her parents were clinically unaffected (Fig. 1A) and had never experienced paralytic symptoms.At admission, she exhibited no abnormal symptoms following physical examination, electrocardiography, and radiological investigation. She had no abnormal findings in laboratory testing (e.g., blood urea nitrogen, 8.4 mg/dL; creatinine, 0.75 mg/dL; Na, 138 mmol/L; K, 4.4 mmol/L; HCO3, 24.5 mmol/L; and Urine, PH 6.0), except for an increased level of serum creatine kinase (621 IU/L; normal range, 32 to 294 IU/L). There was no grip or percussion myotonia. Paramyotonic symptoms, such as muscle stiffness and eyelid lag, were not induced by exposure to cold. Holter monitoring for 24 hours did not reveal any structural abnormalities. In order to induce paralytic symptoms, we administered oral potassium chloride (2.4 g), after which the patient developed paresthesias in both hands after 20 minutes, followed by severe flaccid paralysis of all 4 limbs, with a rating of 1 according to the Medical Research Council scale4). Sensory examination revealed no deficits. Deep tendon reflexes were absent in all limbs, and Babinski and Hoffmann signs were not present. Cognitive and autonomic functions were preserved. No clinical or electrical myotonia was present. During the attack of muscle weakness, the patient showed increased levels of serum potassium (6.5 mEq/L; normal range, 3.5 to 5.0 mEq/L) and creatine kinase (854 IU/L), with the electrocardiograms showing characteristically tall and peaked T waves. Thirty minutes after intravenous injection of glucose and insulin, serum potassium concentrations decreased to 4.2 mEq/L, and both the clinical manifestations and electrocardiographic measurements returned to normal.Initial empirical treatment with acetazolamide (starting at 250 mg twice daily) was administered to the patient, who was also advised to avoid potassium-rich foods, fasting, and strenuous exercise. Prophylactic treatment with acetazolamide was ineffective, however, and the patient was consequently prescribed hydrochlorothiazide (25 mg twice daily). The frequency and severity of paralytic attacks were markedly reduced after administration of this medication. We have been regularly monitoring the serum electrolyte values, including Na, K, and chloride, on an outpatient basis since the patient was discharged from the hospital. The electrolyte levels have remained within normal ranges (e.g., K, 3.7 to 5.0 mEq/L; Na, 136 to 140 mEq/L).Mutation screening was performed by sequence analysis of the entire coding region of the SCN4A gene as has been previously described5). Genomic DNA was extracted from the peripheral blood of the patient and her parents with a DNA extraction kit (SolGent, Daejeon, Korea). All participants provided their written informed consent, and the study was conducted in compliance with the Institutional Review Board of Konyang University Hospital.Direct sequencing of SCN4A exon 13 revealed a heterozygous C>T transition at nucleotide 2111(Fig. 1B), resulting in the substitution of a threonine by a methionine at codon 704 (Thr704Met). This mutation was absent in the patient's parents (Fig. 1B). Parental identity was verified by genotyping 15 short tandem repeat markers by using a PowerPlex 16 system (Promega, Madison, WI, USA).
- Discussion
- Discussion
SCN4A mutations are associated with various neuromuscular disorders, including HYPP, normokalemic periodic paralysis, hypokalemic periodic paralysis, paramyotonia congenita, potassium-aggravated myotonia, and congenital myasthenic syndrome3,6). Despite some overlapping characteristics between these diseases, clinicians have attempted to differentiate between them mainly because the therapeutic response to medications is somewhat different7). In the present study, DNA sequence analysis of the SCN4A gene revealed a de novo Thr704Met mutation in a single member of a family whose parents were clinically unaffected and did not show a mutation in the SCN4A gene. De novo mutations have not previously been reported in HYPP. We think that the reason for this rarity of de novo mutations in HYPP, unlike in other autosomal-dominant channelopathies, is either the low incidence per se or the clinical variability of this disease. Previous studies have revealed that certain SCN4A mutations in HYPP exhibit phenotypic variations8-10). Phenotypic variability may disturb the correlation between genotype and phenotype in HYPP, making it difficult to differentiate the disease from other allelic ion channel pathologies, including paramyotonia congenita, normokalemic periodic paralysis, and potassium-aggravated myotonia. The patient in this case exhibited only flaccid muscle weakness without (para)myotonia, which the authors termed "pure HYPP". A case of pure HYPP with Thr704Met mutation has not previously been reported in Asia.Prophylactic use of acetazolamide has been considered to be highly effective for treating patients with periodic paralyses11-13). Nonetheless, some patients with specific mutations reported no effect and even worsening of paralytic symptoms after acetazolamide treatment14,15). Thiazide diuretics have been used as alternatives to acetazolamide in cases where initial empirical therapy with acetazolamide was ineffective. In the present study, the patient who was unresponsive to acetazolamide reported a marked decrease in the frequency and intensity of paralytic attacks after treatment with hydrochlorothiazide. It remains to be elucidated whether the improvement of paralytic symptoms in our patient is associated with genetic or allelic variations in response to medications.In conclusion, we reported the first de novo SCN4A mutation in a patient with HYPP whose parents were clinically and genetically unaffected. Our findings have implications for the diagnosis and genetic counseling of an isolated family member who has phenotypic features of HYPP. These results also suggest that de novo SCN4A mutations in this disorder may occur more frequently than might previously have been anticipated.
- Acknowledgments
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2008-331-E00158).
- References
- 1. Lehmann-Horn F, Rudel R, Jurkat-Rott K. In: Engel A, Franzini-Armstrong C,Non-dystrophic myotonias and periodic paralyses. editors. Myology: basic and clinical. 20043rd ed. New York: McGraw-Hill, :1257–1300.
[Article]2. Feero WG, Wang J, Barany F, Zhou J, Todorovic SM, Conwit R, et al. Hyperkalemic periodic paralysis: rapid molecular diagnosis and relationship of genotype to phenotype in 12 families. Neurology 1993;43:668–673.
[Article] [PubMed]3. Jurkat-Rott K, Lehmann-Horn F. Genotype-phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics 2007;4:216–224.
[Article] [PubMed]4. Paternostro-Sluga T, Grim-Stieger M, Posch M, Schuhfried O, Vacariu G, Mittermaier C, et al. Reliability and validity of the Medical Research Council (MRC) scale and a modified scale for testing muscle strength in patients with radial palsy. J Rehabil Med 2008;40:665–671.
[Article] [PubMed]5. Park YH, Kim JB. An atypical phenotype of hypokalemic periodic paralysis caused by a mutation in the sodium channel gene SCN4A. Korean J Pediatr 2010;53:909–912.
[Article] [PubMed] [PMC]6. HantaÏ D, Richard P, Koenig J, Eymard B. Congenital myasthenic syndromes. Curr Opin Neurol 2004;17:539–551.
[Article] [PubMed]7. Ricker K, Böhlen R, Rohkamm R. Different effectiveness of tocainide and hydrochlorothiazide in paramyotonia congenita with hyperkalemic episodic paralysis. Neurology 1983;33:1615–1618.
[Article] [PubMed]8. Brancati F, Valente EM, Davies NP, Sarkozy A, Sweeney MG, LoMonaco M, et al. Severe infantile hyperkalaemic periodic paralysis and paramyotonia congenita: broadening the clinical spectrum associated with the T704M mutation in SCN4A. J Neurol Neurosurg Psychiatry 2003;74:1339–1341.
[Article] [PubMed] [PMC]9. Bouhours M, Luce S, Sternberg D, Willer JC, Fontaine B, Tabti N. A1152D mutation of the Na+ channel causes paramyotonia congenita and emphasizes the role of DIII/S4-S5 linker in fast inactivation. J Physiol 2005;565(Pt 2): 415–427.
[Article] [PubMed] [PMC]10. Okuda S, Kanda F, Nishimoto K, Sasaki R, Chihara K. Hyperkalemic periodic paralysis and paramyotonia congenita--a novel sodium channel mutation. J Neurol 2001;248:1003–1004.
[Article] [PubMed]11. Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev 1999;79:1317–1372.
[Article] [PubMed]12. Lehmann-Horn F, Rüdel R. Molecular pathophysiology of voltage-gated ion channels. Rev Physiol Biochem Pharmacol 1996;128:195–268.
[PubMed]13. Ptácek L. The familial periodic paralyses and nondystrophic myotonias. Am J Med 1998;105:58–70.
[Article] [PubMed]
Fig. 1
Family pedigree and mutation analysis. (A) Pedigree of the hyperkalemic periodic paralysis (HYPP) patient shows the proband (indicated by an arrow). (B) Identification of a de novo mutation in the SCN4A gene in the HYPP patient. Electropherograms show the sequence encompassing the heterozygous transition mutation (c.2111C>T) in exon 13 of SCN4A in the patient as well as the corresponding wild-type sequences in normal family members.