 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 60(1); 2017
Malformations of cortical development: genetic mechanisms and diagnostic approach
- Corresponding author: Jeehun Lee, MD, PhD. Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University, School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel: +82-2-3410-0910, Fax: +82-2-3410-0043, jhlee0101@skku.edu
- Received May 29, 2015 Revised November 20, 2015 Accepted December 16, 2015
- Abstract
-
Malformations of cortical development are rare congenital anomalies of the cerebral cortex, wherein patients present with intractable epilepsy and various degrees of developmental delay. Cases show a spectrum of anomalous cortical formations with diverse anatomic and morphological abnormalities, a variety of genetic causes, and different clinical presentations. Brain magnetic resonance imaging has been of great help in determining the exact morphologies of cortical malformations. The hypothetical mechanisms of malformation include interruptions during the formation of cerebral cortex in the form of viral infection, genetic causes, and vascular events. Recent remarkable developments in genetic analysis methods have improved our understanding of these pathological mechanisms. The present review will discuss normal cortical development, the current proposed malformation classifications, and the diagnostic approach for malformations of cortical development.
- Introduction
- Introduction
Malformations of cortical development are rare congenital anomalies of the cerebral cortex that have long been of interest to neuroscientists and pediatricians. The term “malformations of cortical development” was first introduced in 19961,2). Patients with cortical malformations clinically present with epilepsy that is frequently intractable and various degrees of developmental delay and intellectual disability. It is assumed that approximately 40% of children with intractable epilepsy have malformations of cortical development3).Malformations of cortical development encompass a broad spectrum of anomalous cortical formations with diverse anatomical and morphological abnormalities, a variety of genetic causes, and different clinical presentations. The emergence of brain magnetic resonance imaging (MRI) has greatly contributed to the determination of exact morphologies of cortical malformations. The hypothetical mechanisms of cortical malformation include any interruptions during the formation of the cerebral cortex: viral infections, genetic causes, vascular events, etcetera4). Recent developments in genetic analysis methods have improved our understanding of these pathological mechanisms, and offer implications for future therapeutic development. The present review will discuss the process of normal cortical development, the pathogenesis and classification of malformations, and the clinical diagnostic approach for malformations of cortical development.
- Normal cortical development
- Normal cortical development
The development of the cerebral cortex includes a series of complex processes. In the initial step, the proliferation (mitosis) of neuronal precursors in the ventricular zone results in an increase in the number of neurons. Next, neurons migrate from the ventricular zone to the cortex. The last step is the postmigrational organization of the neurons in the cortex5,6).The mature cerebral cortex includes 2 types of cells: cortical neurons and glia. Neurons facilitate the signaling of the nervous system, and glia have supportive roles for the survival and function of neurons. There are 2 types of neurons: excitatory neurons and inhibitory interneurons. Excitatory neurons are born from polarized neuroepithelial cells (neuronal precursors) in the lateral ventricle. Excitatory neurons divide and differentiate into radial glial progenitors that extend fibers to the pial surface (Fig. 1)7). Radial glial progenitors can then divide into intermediate progenitors and outer radial glial progenitors, which divide and differentiate into excitatory projection neurons that migrate along the processes of radial glia to form the cortical plate. The cerebral cortex develops in an “inside-out” fashion. Early-born neurons form the deepest layers (V and VI) and later-born neurons form the more superficial layers of the cortex (II and III). Structural barriers of the pial surface and molecular signals have roles in the arrest of neuronal migration4). In this manner, the 6-layered laminar structure of the cerebral cortex is formed. Inhibitory interneurons are born and migrate tangentially to the dorsal telencephalon, turn and migrate radially to the cortical plate, and are organized with excitatory neurons8,9). Glia are formed from progenitor cells after neurogenesis9,10).
- Pathogenesis and classification of malformations of cortical development
- Pathogenesis and classification of malformations of cortical development
- 1. Group I: malformations secondary to abnormal neuronal and glial proliferation or apoptosis
- 1. Group I: malformations secondary to abnormal neuronal and glial proliferation or apoptosis
1) Microcephaly
1) Microcephaly
Microcephaly is defined by a head circumference less than 2 standard deviations below the average. Primary microcephaly, frequently referred to as microcephaly vera, is characterized by a small brain size without any anomalies in other organs. In primary microcephaly, brain MRI shows a normal structure, although the actual volume of the brain is less than 2 standard deviations below the average (Fig. 2A). Occasionally, a simplified gyral pattern, heterotopia, hypomyelination, or cerebellar hypoplasia can accompany microcephaly4). Microcephaly results from reduced proliferation or accelerated apoptosis. To date, most gene mutations known to cause microcephaly affect neurogenesis pathways by altering regulation of transcription, cell cycle, centrosome duplication and maturation, mitotic spindle disruption, and DNA repair defects (Table 1)2,11,12,13,14,15,16). These factors affect neurogenesis during phases of mitosis, which can produce proliferation abnormalities and result in a smaller population of neurons.2) Megalencephaly
2) Megalencephaly
Megalencephaly refers to a condition in which the brain is more than 2 standard deviations above average (Fig. 2B). This group is not fully defined, and 6% of patients with polymicrogyria show megalencephaly17).The discovery of specific pathways that control cell proliferation has informed the pathogenesis of several malformations of cortical development. The mammalian target of rapamycin (mTOR) pathway plays an important role in abnormal cortical development of the tuberous sclerosis complex (TSC)18). The discovery of the mTOR pathway's role in TSC has had great therapeutic implications for TSC: mTOR pathway inhibitors are now used in the treatment of subependymal giant cell astrocytoma19).3) Hemimegalencephaly
3) Hemimegalencephaly
Hemimegalencephaly is the overgrowth of one brain hemisphere or part of a hemisphere. The pathology typically reveals various degrees of cortical dysplasia, white matter abnormalities, and polymicrogyria. Hemimegalencephaly can present as an isolated anomaly or it can exist as part of syndrome such as Klippel-Trenauney syndrome or hypomelanosis of Ito20). Patients with hemimegalencephaly present with mental retardation, early-onset intractable epilepsy, and contralateral hemiparesis. Brain MRI of these patients reveals abnormal enlargement of at least one lobe or one hemisphere, white matter signal abnormalities, pachygyria, and heterotopia (Fig. 3A)4). De novo somatic mutation of the AKT3 pathway was proposed to be associated with hemimegalencephaly in surgical specimens21,22).4) Focal cortical dysplasia
4) Focal cortical dysplasia
Focal cortical dysplasia is one of the most frequent causes of intractable focal epilepsy. The pathology shows dysplastic neurons, balloon cells, and cortical dyslamination23,24). High resolution MRI can identify blurring of the gray-white matter junction, abnormal gyral thickening, and gray or white matter abnormalities (Fig. 3B)24,25). The pathogenesis of focal cortical dysplasia has yet to be revealed; however, brain somatic mutation of mTOR was recently proposed to underlie type II focal cortical dysplasia26).
Group I is associated with interruptions in the initial stages of neuronal precursor proliferation as well as disequilibrium between the proliferation and apoptosis of neuronal precursors. Subgroups include (1) group I.A: microcephaly; (2) group I.B: megalencephaly; and (3) group I.C: cortical dysgenesis with abnormal cell proliferation.- 2. Group II: malformations due to abnormal neuronal migration
- 2. Group II: malformations due to abnormal neuronal migration
1) Heterotopia (group II.A)
1) Heterotopia (group II.A)
Periventricular heterotopia is characterized by neuronal nodules in the periventricular area and a normal cerebral cortex. Heterotopia results from the failure to initiate migration in a small group of neurons. Pathological specimens show normal-shaped neurons and glia accompanied by myelinated fibers and gliosis27). Approximately 90% of patients present with various types of seizures, mostly in adolescence. Brain MRI of these patients shows variously sized nodules along the lateral ventricles (Fig. 4A). The possible causes of periventricular heterotopia are genetic2) Classical (type I) lissencephaly (group II.B)
2) Classical (type I) lissencephaly (group II.B)
Lissencephaly is one of the best-known malformations of cortical development, referred to as “smooth brain,” and results from the absence of normal gyri and sulci formation. Patients with type I lissencephaly have severe intellectual disability, microcephaly, and intractable epilepsy including infantile spasms. There are several genes that have been identified as causal to this malformation, including LIS1, DCX, TUBA1A, ARX, and RELN (Table 1)28,29,30,31). Brain MRI of these patients shows snowman-shaped configurations with areas of pachygyria and agyria (Fig. 4B). The posterior head region is more severely affected in patients with LIS1 mutations, while the frontal and temporal areas are smoother in patients with DCX mutations32).3) Subcortical band heterotopia (group II.C)
3) Subcortical band heterotopia (group II.C)
Subcortical band heterotopia is also referred to as “double cortex” syndrome, because it is characterized by a band of subcortical heterotopic neurons between the ventricle and cerebral cortex. This disorder is mainly seen in females and is associated with intellectual disability and intractable epilepsy. Subcortical band heterotopia is known to be associated with mutations of the DCX gene33). The function of the DCX protein is to direct neuronal migration by regulating the organization and stability of microtubules that facilitate neuronal motility during cortico-genesis28). Mutation of the DCX gene in males causes classical lissencephaly. Brain MRI of these patients reveals a thin layer of white matter between 2 layers of gray matter (Fig. 4C).4) Cobble stone malformations (type II lissencephaly, group II.D)
4) Cobble stone malformations (type II lissencephaly, group II.D)
Type II lissencephaly is characterized by the nodular appearance of the cerebral cortex due to disorganization of the cortical layers, and excessive migration of neurons through the pial surface. This disorder is frequently associated with various eye abnormalities and congenital muscular dystrophies4). Clinically, increases in serum creatine kinase in patients with type II lissencephaly can expedite diagnosis. The clinical phenotype varies according to the causative genes. Fukuyama muscular dystrophy is the mildest form of type II lissencephaly, mainly existing in Japan and rarely in Korea (Fig. 5A)34,35). Muscle-eye-brain disease has been mainly reported in Finnish populations with early onset eye symptoms, intellectual disability, myoclonus, and congenital muscular dystrophy36). Walker–Warburg syndrome is the most severe form of type II lissencephaly and results in early infantile death37).
Group II is divided into 4 subgroups according to the timing of neuronal migration interruption: (1) group II.A, periventricular heterotopia (problems in the initiation of migration); (2) group II.B, generalized abnormalities of transmantle migration (lissencephalies); (3) group II.C, localized abnormalities of transmantle migration (subcortical heterotopia); and (4) group II.D, anomalies associated with abnormal terminal migration or defects in pial limiting membranes (cobblestone malformations)2).- 3. Group III: malformations secondary to abnormal postmigrational development
- 3. Group III: malformations secondary to abnormal postmigrational development
1) Polymicrogyria and schizencephaly (group III.A)
1) Polymicrogyria and schizencephaly (group III.A)
Polymicrogyria refers to a lumpy appearance of the cerebral cortex resultant from numerous small gyri separated by shallow sulci. This malformation can be focal or diffuse, and unilateral or bilateral. Bilateral polymicrogyria is frequently seen affecting the frontal, fronto-parietal, parieto-occipital, perisylvian, or medial occipital areas. Polymicrogyria is associated with various degrees of intellectual disability, motor dysfunction, and epilepsy. Familial polymicrogyria syndrome involves the fronto-parietal and perisylvian areas and is reported to be associated with mutation of GPR5638). Brain MRI of these patients shows small irregular gyri and vague gray/white matter differentiation.Schizencephaly is characterized by the appearance of a cleft between the cortex and ventricle that is lined by polymicrogyric cortex (Fig. 5B). This malformation can be unilateral and bilateral; type I includes “closed-lip” clefts and type II includes “open-lip” clefts. The phenotype is more severe in type II, with patients showing developmental delay, microcephaly, intractable epilepsy, and motor deficits in the contralateral limbs4).2) Focal cortical dysplasia (group III.C)
2) Focal cortical dysplasia (group III.C)
Focal cortical dysplasia is the most common cause of intractable focal epilepsy and is frequently considered a candidate for epilepsy surgery. Certain forms of focal cortical dysplasia are classified as group III because they can result from injury to the cerebral cortex in the later stages of cerebral cortical development. There have been reports about the various causes of mild forms of focal cortical development (type I), and these include prenatal and perinatal injuries such as severe prematurity, asphyxia, bleeding, hydrocephalus, and stroke39,40). In addition, group III.C cases are associated with physical injury, vascular malformation, or epileptogenic tumor by definition.
According to the classifications proposed in 2012, polymicrogyria is divided into 4 groups: group III.A, with schizencephalic clefts or calcifications that are presumably due to infection or vascular causes; group III.B, without clefts or calcifications, and may be genetic or disruptive; group III.C, as part of genetically defined multiple congenital anomaly syndromes; and group III.D, which is associated with inborn errors of metabolism2).
Malformations of cortical development result from the disruption of normal cortical development processes. In 1996, the initial classification scheme for cortical development was introduced based on the stage in which the developmental process was disturbed1). This scheme has since been updated and revised according to newly discovered mechanisms of pathogenesis; the most recent revision was published in 20122). There are 3 groups of cortical development malformation based on the timing and pathogenesis of the disruption: (1) group I, malformations secondary to abnormal neuronal and glial proliferation or apoptosis; (2) group II, malformations due to abnormal neuronal migration; and (3) group III, malformations associated with abnormal postmigrational development.
- Diagnostic approach to malformations of cortical development
- Diagnostic approach to malformations of cortical development
Patients with malformations of cortical development usually present with intractable seizures or developmental delay. The phenotype is extremely heterogeneous; the age of presentation, the degree of developmental delay, and the severity of neurologic deficit can vary according to the type of malformation. Clinicians diagnose malformations of cortical development following brain MRI as a diagnostic process for seizure or developmental delay. Then, a radiologic classification is made according to the morphological characteristics of the brain MRI. The diagnostic steps are summarized in Fig. 6. The dysmorphisms and anomalies accompanying cortical malformations require thorough investigation. If brain MRI findings or phenotype imply a certain genetic disorder such as periventricular nodular heterotopia, lissencephaly, or cobble stone malformations, Sanger sequencing for the candidate gene is the most direct method of diagnosis. If there are associated anomalies or dysmorphisms such as Miller-Dieker syndrome, a chromosome study or chromosomal microarray is necessary. If there are multiple candidate genes for a given cortical malformation group, such as microcephaly without other specific phenotype, targeted gene panel testing using next generation sequencing is more useful. If no causative gene is found by targeted gene sequencing, trio whole-exome sequencing for the proband and parents can be considered for a genetic diagnosis. Genetic diagnosis is recommended because it is a prerequisite for genetic counseling, explanation of the disease course, and prognosis. At present, Sanger sequencing for the causative genes of malformations of cortical dysplasia and targeted gene sequencing are not formally permitted by the Korean National Health Insurance Service. However, most genetic analyses are possible on a research basis, and these tests will be permitted formally in the near future.
- Conclusion
- Conclusion
Research investigating the genetic basis of various malformations of cortical development has improved our understandings of brain development and the pathophysiological basis of cortical malformations. Different mutations of the same gene can cause different phenotypic presentations depending on the degree of protein dysfunction. Loss-of-function mutations or gain-of-function mutations cause different phenotypes. Mosaicism can occur, revealing focal or milder phenotypic variations. In addition, functional mosaicism occurs due to X chromosome inactivation. In cases of gene mutation, causative management is not possible; however, elucidating causative genes and pathological mechanisms is a great step towards therapeutic development.Malformations of cortical development have become increasingly recognized with the introduction of brain MRI. Cortical malformations are a major cause of intractable epilepsy and developmental delay. Genetic mutations and the resultant pathologies explain some malformations of cortical development, and these mechanisms are mainly associated with neuronal precursor proliferation, migration, and organization. Research about the pathogenesis of cortical malformation continues to elucidate the secrets of cerebral development. Diagnosis of malformations of cortical development will lead to better treatment for seizures and developmental delay, and contribute to genetic counseling for the family. Future studies of the pathogenesis of malformations of cortical development will enhance the development of treatments for various stages of brain development.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Barkovich AJ, Kuzniecky RI, Dobyns WB, Jackson GD, Becker LE, Evrard P. A classification scheme for malformations of cortical development. Neuropediatrics 1996;27:59–63.
[Article] [PubMed]2. Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 2012;135(Pt 5): 1348–1369.
[Article] [PubMed] [PMC]3. Kuzniecky RI. Magnetic resonance imaging in developmental disorders of the cerebral cortex. Epilepsia 1994;35(Suppl 6): S44–S56.
[Article] [PubMed]4. Pang T, Atefy R, Sheen V. Malformations of cortical development. Neurologist 2008;14:181–191.
[Article] [PubMed] [PMC]5. Chenn A, McConnell SK. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 1995;82:631–641.
[Article] [PubMed]6. Hu WF, Chahrour MH, Walsh CA. The diverse genetic landscape of neurodevelopmental disorders. Annu Rev Genomics Hum Genet 2014;15:195–213.
[Article] [PubMed] [PMC]7. Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell 2011;146:18–36.
[Article] [PubMed] [PMC]8. Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci 2006;7:687–696.
[Article] [PubMed]9. Reillo I, de Juan Romero C, García-Cabezas MÁ, Borrell V. A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb Cortex 2011;21:1674–1694.
[Article] [PubMed]10. Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci 2013;14:755–769.
[Article] [PubMed]11. Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet 2009;25:501–510.
[Article] [PubMed] [PMC]12. Desir J, Cassart M, David P, Van Bogaert P, Abramowicz M. Primary microcephaly with ASPM mutation shows simplified cortical gyration with antero-posterior gradient pre- and post-natally. Am J Med Genet A 2008;146A:1439–1443.
[Article] [PubMed]13. Yu TW, Mochida GH, Tischfield DJ, Sgaier SK, Flores-Sarnat L, Sergi CM, et al. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet 2010;42:1015–1020.
[Article] [PubMed] [PMC]14. Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 2004;44:279–293.
[Article] [PubMed]15. Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet 2010;42:245–249.
[Article] [PubMed] [PMC]16. Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet 2008;40:232–236.
[Article] [PubMed]17. Leventer RJ, Jansen A, Pilz DT, Stoodley N, Marini C, Dubeau F, et al. Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients. Brain 2010;133(Pt 5): 1415–1427.
[Article] [PubMed] [PMC]18. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345–1356.
[Article] [PubMed]19. de Vries PJ. Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics 2010;7:275–282.
[Article] [PubMed] [PMC]20. Tinkle BT, Schorry EK, Franz DN, Crone KR, Saal HM. Epidemiology of hemimegalencephaly: a case series and review. Am J Med Genet A 2005;139:204–211.
[PubMed]21. Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012;74:41–48.
[Article] [PubMed] [PMC]22. Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012;44:941–945.
[Article] [PubMed] [PMC]23. Fauser S, Huppertz HJ, Bast T, Strobl K, Pantazis G, Altenmueller DM, et al. Clinical characteristics in focal cortical dysplasia: a retrospective evaluation in a series of 120 patients. Brain 2006;129(Pt 7): 1907–1916.
[Article] [PubMed]24. Blümcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–174.
[Article] [PubMed]25. Tassi L, Colombo N, Garbelli R, Francione S, Lo Russo G, Mai R, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 2002;125(Pt 8): 1719–1732.
[Article] [PubMed]26. Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015;21:395–400.
[Article] [PubMed]27. Tassi L, Colombo N, Cossu M, Mai R, Francione S, Lo Russo G, et al. Electroclinical, MRI and neuropathological study of 10 patients with nodular heterotopia, with surgical outcomes. Brain 2005;128(Pt 2): 321–337.
[Article] [PubMed]28. Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 1998;92:63–72.
[Article] [PubMed]29. Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 1993;364:717–721.
[Article] [PubMed]30. Poirier K, Keays DA, Francis F, Saillour Y, Bahi N, Manouvrier S, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum Mutat 2007;28:1055–1064.
[Article] [PubMed]31. Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, Cleak J, et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 2007;128:45–57.
[Article] [PubMed] [PMC]32. Dobyns WB, Berry-Kravis E, Havernick NJ, Holden KR, Viskochil D. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet 1999;86:331–337.
[Article] [PubMed]33. des Portes V, Pinard JM, Smadja D, Motte J, Boespflüg-Tanguy O, Moutard ML, et al. Dominant X linked subcortical laminar heterotopia and lissencephaly syndrome (XSCLH/LIS): evidence for the occurrence of mutation in males and mapping of a potential locus in Xq22. J Med Genet 1997;34:177–183.
[Article] [PubMed] [PMC]34. Fukuyama Y, Osawa M, Suzuki H. Congenital progressive muscular dystrophy of the Fukuyama type - clinical, genetic and pathological considerations. Brain Dev 1981;3:1–29.
[Article] [PubMed]35. Lee J, Lee BL, Lee M, Kim JH, Kim JW, Ki CS. Clinical and genetic analysis of a Korean patient with Fukuyama congenital muscular dystrophy. J Neurol Sci 2009;281:122–124.
[Article] [PubMed]36. Santavuori P, Somer H, Sainio K, Rapola J, Kruus S, Nikitin T, et al. Muscle-eye-brain disease (MEB). Brain Dev 1989;11:147–153.
[Article] [PubMed]37. Dobyns WB, Pagon RA, Armstrong D, Curry CJ, Greenberg F, Grix A, et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet 1989;32:195–210.
[Article] [PubMed]38. Piao X, Chang BS, Bodell A, Woods K, Benzeev B, Topcu M, et al. Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol 2005;58:680–687.
[Article] [PubMed]39. Marín-Padilla M, Parisi JE, Armstrong DL, Sargent SK, Kaplan JA. Shaken infant syndrome: developmental neuropathology, progressive cortical dysplasia, and epilepsy. Acta Neuropathol 2002;103:321–332.
[Article] [PubMed]40. Krsek P, Jahodova A, Maton B, Jayakar P, Dean P, Korman B, et al. Low-grade focal cortical dysplasia is associated with prenatal and perinatal brain injury. Epilepsia 2010;51:2440–2448.
[Article] [PubMed]41. Trimborn M, Bell SM, Felix C, Rashid Y, Jafri H, Griffiths PD, et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am J Hum Genet 2004;75:261–266.
[Article] [PubMed] [PMC]42. Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet 2002;32:316–320.
[Article] [PubMed]43. Hung LY, Tang CJ, Tang TK. Protein 4.1 R-135 interacts with a novel centrosomal protein (CPAP) which is associated with the gamma-tubulin complex. Mol Cell Biol 2000;20:7813–7825.
[Article] [PubMed] [PMC]44. Bhat V, Girimaji SC, Mohan G, Arvinda HR, Singhmar P, Duvvari MR, et al. Mutations in WDR62, encoding a centrosomal and nuclear protein, in Indian primary microcephaly families with cortical malformations. Clin Genet 2011;80:532–540.
[Article] [PubMed]45. Adachi H, Tsujimoto M, Hattori M, Arai H, Inoue K. cDNA cloning of human cytosolic platelet-activating factor acetylhydrolase gamma-subunit and its mRNA expression in human tissues. Biochem Biophys Res Commun 1995;214:180–187.
[Article] [PubMed]46. Zaki M, Shehab M, El-Aleem AA, Abdel-Salam G, Koeller HB, Ilkin Y, et al. Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am J Med Genet A 2007;143A:939–944.
[Article] [PubMed]47. Leandro-García LJ, Leskelä S, Landa I, Montero-Conde C, López-Jiménez E, Letón R, et al. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken) 2010;67:214–223.
[Article] [PubMed]48. Fox JW, Lamperti ED, Ekşioğlu YZ, Hong SE, Feng Y, Graham DA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 1998;21:1315–1325.
[Article] [PubMed]49. Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 2002;11:981–991.
[Article] [PubMed]50. Hewitt JE. Abnormal glycosylation of dystroglycan in human genetic disease. Biochim Biophys Acta 2009;1792:853–861.
[Article] [PubMed]51. Raducu M, Baets J, Fano O, Van Coster R, Cruces J. Promoter alteration causes transcriptional repression of the POMGNT1 gene in limb-girdle muscular dystrophy type 2O. Eur J Hum Genet 2012;20:945–952.
[Article] [PubMed] [PMC]52. Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 2013;45:639–647.
[Article] [PubMed]53. Jurado LA, Coloma A, Cruces J. Identification of a human homolog of the Drosophila rotated abdomen gene (POMT1) encoding a putative protein O-mannosyl-transferase, and assignment to human chromosome 9q34.1. Genomics 1999;58:171–180.
[Article] [PubMed]
Fig. 1
Replication of neuronal precursors and formation of the neocortex. (A) Neuroepithelial cells divide into new neuroepithelial cells, radial glia, intermediate progenitors, and outer radial glia. (B) Outer radial glia replicate and also form migrating neurons. Migrating neurons make the cortical layers in an inside-out fashion.
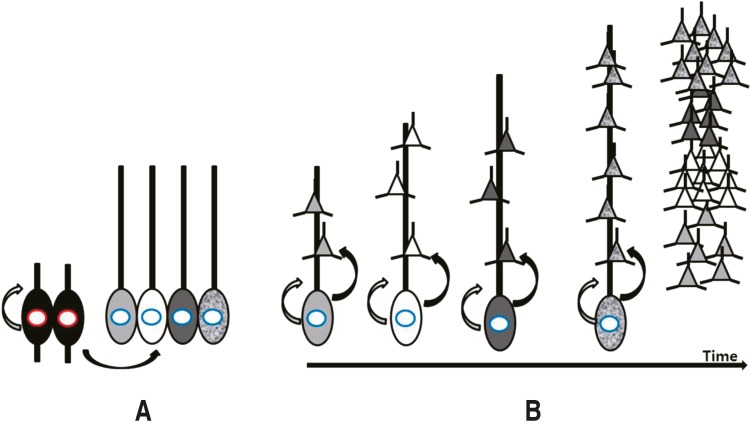
Fig. 2
Microcephaly and megalencephaly. (A) Brain MRI of an 18-month-old male patient revealing a decrease in brain volume as compared to the size of face with relative preservation of normal structure. The patient's head circumference was below the 3rd percentile. (B) Brain MRI of a 3-year-old male patient with global developmental delay showing a relatively larger head size as compared to the size of the face, and mild ventriculomegaly without structural anomaly.
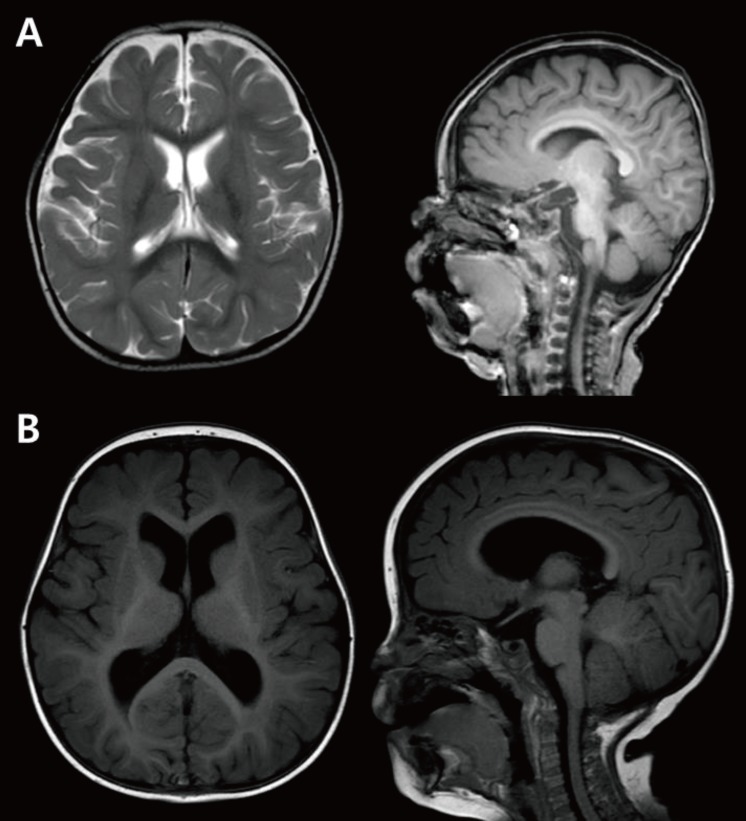
Fig. 3
Hemimegalencephaly and focal cortical dysplasia type II. (A) Brain magnetic resonance imaging of a 2-year-old male patient with global developmental delay showing left hemispheric enlargement accompanying cortical thickening and a high white matter signal intensity in T2-weighted imaging. (B) The T2-weighted and coronal images of a 5-year-old male patient with intractable partial seizures showing thickening of the cortex (long arrow) with abnormal white matter signal (short arrow).
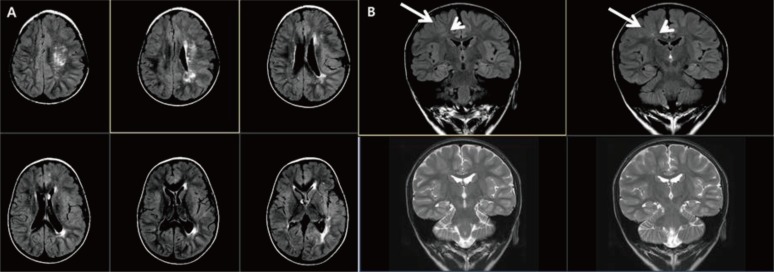
Fig. 4
Neuronal migration anomaly. (A) A 1-month-old female patient showing bilateral nodular heteretopia with gray matter signal round nodules in the periventricular areas. (B) Miller-Dieker syndrome. A neonate with Fallot tetralogy, hypospadias, and ventriculomegaly in uterine ultrasonography revealing lissencephaly, and with a deletion in chromosome 17p13.3. (C) Brain magnetic resonance imaging of a 15-year-old female patient with a DCX gene point mutation (c.1051+2T>A) showing subcortical band heterotopia between thin cortex and the ventricle.
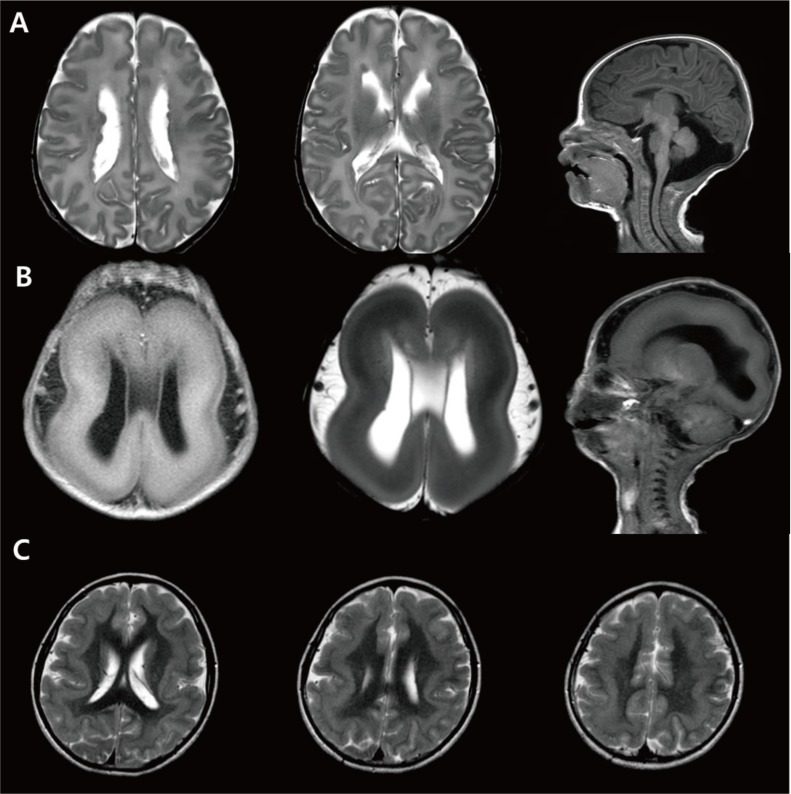
Fig. 5
Polymicrogyria and schizencephaly. (A) Brain magnetic resonance imaging of a 10-year-old male patient with global developmental delay and increased serum creatine kinase showing cortical thickening (arrows). (B) A 13-month-old male patient with left hemiplegia and global developmental delay showing bilateral open-lip schizencephaly and polymicrogyric cortex near the lining of the separated cortex (arrows).
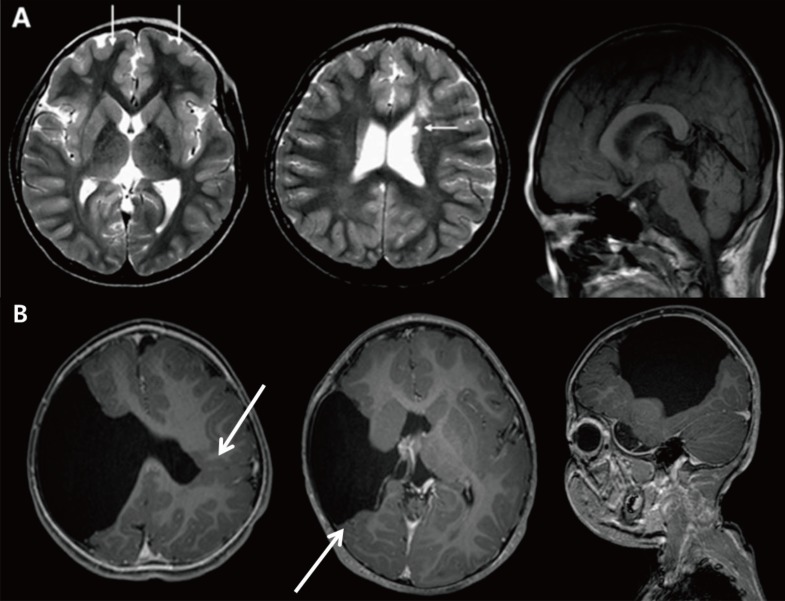
Fig. 6
Proposed steps for the genetic diagnosis of malformations of cortical development. USG, ultrasonography.

Table 1
The representative genes associated with various malformations of cortical development
| Gene | MIM number | Known function | Phenotypes |
|---|---|---|---|
| Microcephaly | |||
| MCPH1 | *607117 | Encodes a regulator of chromosome condensation41) | Microcephaly 1 (AR) |
| ASPM | *605481 | Encodes gene which is essential for normal mitotic spindle function in embryonic neuroblasts42) | Microcephaly 5 (AR) |
| CENPJ | *609279 | Encodes a centrosomal protein with a putative role in regulation of microtubule assembly and nucleation43) | Microcephaly 6 (AR) |
| WDR62 | *613583 | Encodes a protein that localizes to the centrosome and to the nucleus, depending on the cell phase and on the cell type44) | Microcephaly 2 (AR) with or without cortical malformation |
| Abnormal neuronal migration | |||
| PAFAH1B1 | *601545 | NDEL1 interacts with LIS1 to sustain the function of dynein, which in turn impacts microtubule organization, nuclear translocation, and neuronal positioning45) | Lissencephaly 1, subcortical band heterotopia |
| DCX | *300121 | Microtubule associated protein, stabilizes microtubules against depolymerization32) | X-linked Lissencephaly, X-linked subcortical laminar heterotopia |
| TUBA1A | *602529 | Encodes neuronal alpha1-tubulin32) | Lissencephaly 3; microcephaly and pachygyria; thick gyri |
| NDE1 | *609449 | Encodes a protein with a role in mitosis; NDE1 and LIS1 (*601545) interact and are involved in cerebral cortical development14) | Lissencephaly 4 (with microcephaly) |
| RELN | *600514 | Encodes reelin, a large secreted glycoprotein that activates a signaling pathway in postmitotic migrating neurons required for proper positioning of neurons within laminated nervous system parenchyma46) | Lissencephaly 2 (AR) |
| TUBB3 | *602661 | Neuronal beta-tubulin isoforms47) | Cortical dysplasia, complex, with other brain malformations 1 (AD) |
| FLNA | *300017 | Actin-binding protein that regulates reorganization of the actin cytoskeleton by interacting with integrins, transmembrane receptor complexes, and second messengers48) | Periventricular heterotopia (XLD) |
| ARX | *300382 | Encodes the Aristaless-related homeobox protein, which belongs to the Aristalessrelated subset of the paired class of homeodomain proteins, which play crucial roles in cerebral development and patterning49) | Lissencephaly, infantile spasms, intellectual disability |
| Abnormal postmigrational development | |||
| FKRP | *606596 | Suggest a role in protein glycosylation, their enzyme activities have not yet been defined50) | Muscular dystrophy-dystroglycanopathy type A, B, C, 5 |
| POMGnT1 | *606822 | O-mannose beta-1,2-N-acetylglucosaminyltransferase, participates in O-mannosyl glycan synthesis51) | Muscular dystrophy-dystroglycanopathy type A, B, C, 3 |
| DYNC1H1 | *600112 | Dynein heavy-chain isoform52) | Microcephaly and pachygyria |
| POMT1 | *607423 | Encodes protein O-mannosyltransferase, an enzyme that catalyzes O-mannosylation of proteins53) | Muscular dystrophy-dystroglycanopathy type A, B, C, 1 |
| POMT2 | *607439 | Catalyze the first step in the synthesis of the O-mannosyl glycan found on alphadystroglycan54) | Muscular dystrophy-dystroglycanopathy type A, B, C, 2 |
| LAMA1 | *150320 | Basement membrane protein composed of 3 nonidentical chains arranged in a cross-shaped structure55) | Posterior predominant COB, with congenital muscular dystrophy |
MIM, Mendelian Inheritance in Man (http://www.omim.org); AR, autosomal recessive; AD, autosomal dominant; XLD, X-linked dominant; COB, cobblestone malformation complex.
*This table is based on the information and description of Online Mendelian Inheritance in Man database.