About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 60(3); 2017
A compound heterozygous mutation in the FMO3 gene: the first pediatric case causes fish odor syndrome in Korea
- Corresponding author: Ji Hyun Kim, MD. Division of Endocrinology and Metabolism, Department of Pediatrics, Dongguk University Ilsan Hospital, Dongguk University College of Medicine, 27, Dongguk-ro, Ilsandong-gu, Goyang 10326, Korea. Tel: +82-31-961-7190, Fax: +82-31-961-7188, eogurdl@gmail.com
- Received August 05, 2016 Revised February 10, 2017 Accepted February 16, 2017
- Abstract
-
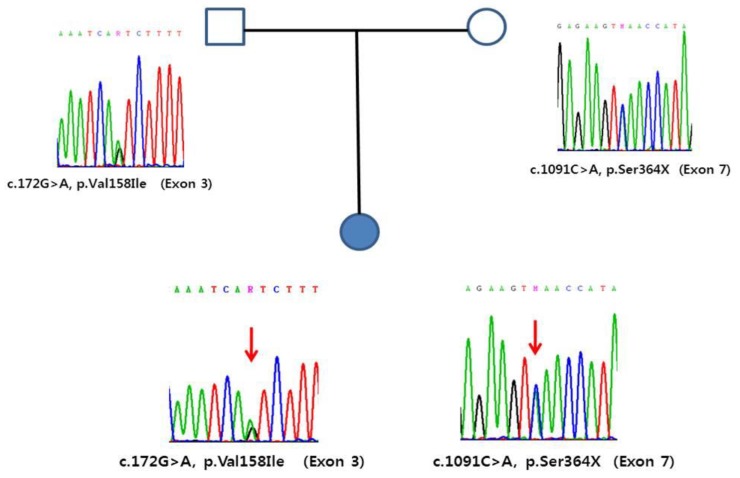
Trimethylaminuria (TMAuria), known as “fish odor syndrome,” is a congenital metabolic disorder characterized by an odor resembling that of rotting fish. This odor is caused by the secretion of trimethylamine (TMA) in the breath, sweat, and body secretions and the excretion of TMA along with urine. TMAuria is an autosomal recessive disorder caused by mutations in flavin-containing monooxygenase 3 (FMO3). Most TMAuria cases are caused by missense mutations, but nonsense mutations have also been reported in these cases. Here, we describe the identification of a novel FMO3 gene mutation in a patient with TMAuria and her family. A 3-year-old girl presented with a strong corporal odor after ingesting fish. Genomic DNA sequence analysis revealed that she had compound heterozygous FMO3 mutations; One mutation was the missense mutation p.Val158Ile in exon 3, and the other was a novel nonsense mutation, p.Ser364X, in exon 7 of the FMO3 gene. Familial genetic analyses showed that the p.Val158Ile mutation was derived from the same allele in the father, and the p.Ser364X mutation was derived from the mother. This is the first description of the p.Ser364X mutation, and the first report of a Korean patient with TMAuria caused by novel compound heterozygous mutations.
- Introduction
- Introduction
Trimethylaminuria (TMAuria) known as “fish odor syndrome,” is a rare metabolic disorder characterized by excessive trimethylamine (TMA) in body secretions. The disorder is due to defects in regulation of the biotransformation pathway that converts malodorous TMA into nonodorous trimethylamine N-oxide (TMAO). Consequently, patients exude an unpleasant body odor resembling that of corruption fish and suffer social and psychological problems. Primary TMAuria is an inherited disorder caused by mutations in the flavin-containing monooxygenase 3 (FMO3) gene. More than 30 mutations have been identified that severely reduce FMO3 activity. Persistent TMAuria is autosomally recessively inherited, and the majority of pathogenic mutations described are missense mutations. Secondary TMAuria is due to TMA or to TMA-precursor overload. Some secondary forms are acquired as a consequence of viral hepatitis or of disease states during adult life. Others are transient, being associated with menstruation. Herein, we report a Korean girl who had a novel causative mutation in the FMO3 gene.
- Case report
- Case report
A 3-year-old girl presented with a chronic strong corporal scent after eating fish. Her birth weight was 2.52 kg, she had no perinatal problems, and had no specific medical history. One to 2 years ago, after eating fish, the dried fish smells and the symptoms become worse with fever a week before the visit. Her parents did not show such symptoms. A physical examination revealed height of 94.8 cm (−0.57 standard deviation score [SDS]) and weight of 14 kg (−0.44 SDS). Her growth and development were normal. TMAuria was suspected from the history, so we performed confirmative genetic testing. The complete blood cell, blood urea nitrogen, creatinine, aspartate aminotransferase, alanine aminotransferase, uric acid and urinalysis showed normal values, and we did not conduct a provocation test using gas chromatography-mass spectrometry because of the fastidious test methods.Genomic DNA was extracted from peripheral blood leukocytes using a QIAamp DNA Blood Midi Kit according to the manufacturer's instructions (Qiagen, Valencia, CA, USA). Direct sequencing of the entire coding exons and flanking intronic sequences of the FMO3 gene was performed using primer pairs designed by the authors. Polymerase chain reaction amplification was performed in a thermal cycler (Model 9700; Applied Biosystems, Foster City, CA, USA) and cycle sequencing was performed on an ABI Prism 3730xl DNA Analyzer using the BigDye Terminator Sequencing Ready Reaction Kit (Applied Biosystems). Sequence variations were analyzed via comparison with the reference sequence. Compound heterozygous variants of c.172G>A (p.Val158Ile) and c.1091C>A (p.Ser364X) were detected in the patient. Additional sequencing for parents indicated that variants came from each parents. A genomic DNA sequence analysis revealed a compound heterozygous mutation; one was a missense mutation for p.Val158Ile in exon 3 and the other was a nonsense mutation for p.Ser364X in exon 7 of the FMO3 gene. A familial genetic analysis showed that the p.Val158Ile was derived from father and that p.Ser364X was derived from the mother (Fig. 1). The p.Ser364X is a novel mutation. The parents of patient felt decreasing body odor on a choline and TMAO-restricted diet, such as a milk, eggs, beans, peanuts, and seafood.
- Discussion
- Discussion
TMAuria, also known as the “fish odor syndrome, ” is a rare metabolic disorder characterized by excessive excretion of TMA in the urine, sweat, breath, and other body secretions1). The defect is due to a failure in the regulation of the biotransformation pathway that converts malodorous TMA into nonodorous TMAO. In humans, TMAO is ingested when marine fish are eaten, which is reduced to TMA by colonic microflora, and is absorbed by passive diffusion across the cell membranes. TMA enters the enterohepatic circulation and is oxygenated to odorless TMAO by FMO3 in normal liver cells. TMAO is very water soluble and is mainly excreted in urine in normal subjects. TMA is produced from bacterial degradation of dietary choline, carnitine, and lecithin found in eggs, beans, and saltwater fish and is converted to nonodorous TMAO by FMO32,3).The disorder is present from birth, but it manifests when high amounts of choline or TMAO are introduced into the diet4). More than 200 cases of this condition have been described worldwide5), and this figure is almost certainly an underestimate.Two major forms of TMAuria have been reported. The primary genetic form causes decreased FMO3 activity, and a secondary form is due to TMA or to TMA-precursor overload. A cases of TMAuria reported in Korea in the past was secondary form associated with chronic viral hepatitis6). The main function of FMOs is to catalyze the conversion of lipophilic compounds to more polar, oxygenated, readily excreted metabolites7). The diagnosis is made on the basis of the clinical presentation and urinalysis. Urine can be analyzed for the concentration of both TMA and TMAO, and the results may be given as an oxidizing ratio; TMAO/(TMAO+ TMA)×100%. TMAuria should have a ratio of greater than 92%. Because it is critical to ingest adequate substrate to maximize the result, some loading tests fail on occasion to provoke satisfactory excretion of metabolic products. Genetic testing can identify individuals affected by the severe primary genetic form of the disorder and distinguish these form those affected by milder genetic forms and those whose symptoms are not due to genetic factors. Thus confirmatory genetic study may improve understanding and treatment of patients with a suggestive history of symptoms. Persistent TMAuria is caused by an autosomal recessively inherited impairment of hepatic TMA oxidation due to a FMO3 deficiency. The primary genetic form of the disease comprises most of the reported cases, and clinical symptoms are confirmed genetically to be due to inactivating mutations. Up to now 63 distinct pathogenic variants have been reported. The majority of pathogenic mutations described are missense mutations8). Nonsense mutations, small deletions resulting in frame-shift mutations, and one large deletion have also been reported. Patients who are homozygous or compound heterozygous mutations severely impair FMO3 activity and decreased enzyme activity by about 50%. Also TMAuria may be caused some representative ethnic-specific FMO3 mutations. The p.Pro153Leu or p.Glu305Ter variants have been reported in Caucasians and p.Cys197Ter, p.Arg205Cys, and p.Arg500Ter FMO3 alleles have been found in a Japanese cohort. The Pro153Leu, Glu305Ter, Cys197Ter and Arg500Ter FMO3 proteins had no detectable FMO3 proteins activity5). The p.Val158Ile, which was one of the mutations found in our patient, has been also reported in Thai and Japanese subjects9). The catalytic efficiencies with Val158Ile, Arg205Cys FMO3 proteins were moderately decreased compared to the wild type protein5). In general, the greater the reduction in FMO3 enzyme activity, the greater severity of symptoms and the less response to therapy.TMAuria is not associated with mortality or morbidity, but the psychosocial aspects of the condition can be devastating10). Patients usually present in childhood or early adulthood, complaining of body odor. Although it may appear to be a small medical problem, sufferers always comment on the major social effects, with consequences for self-esteem, employment, social isolation, and mood disorders, and rare cases of attempted suicide1,11). This condition can be particularly severe for young children and adolescents, who may suffer from scorn, lose confidence, and may stop attending school12).Dietary treatment is the main therapeutic approach, with restrictions on TMA, such as milk obtained from wheat-fed cows, TMA precursors, including choline, eggs, liver, kidney, peas, beans, peanuts, and soya products, and TMAO-containing fish, such as cephalopods and crustaceans13). This is particularly true of “mild” or moderate forms of primary TMAuria. Affected individuals respond differently to different forms of dietary restriction. The use of soaps and body lotions with a pH close to that of normal skin (pH 5.5–6.5) helps retain secreted TMA in a less volatile salt form that can be removed by washing.Another treatment, which exerts its effect in the gastrointestinal tract, is the use of antimicrobials. Short courses of oral neomycin, metronidazole, and amoxicillin have been reported to be useful in some cases. The mechanism involves the destruction of gut bacteria, which are responsible for the reduction of TMAO into TMA. However, antimicrobial therapy presents a number of possible side effects if used chronically, so this treatment option is best suited for transient cases14). Such treatment may be useful when dietary restriction needs to be relaxed, or when TMA production appears to increase (e.g., during menstruation, infection, emotional upset, stress, or exercise). Three antibiotics with different target organisms have been used: metronidazole, amoxicillin, and neomycin. Neomycin appears to be the most effective in preventing formation of TMA from choline4). Supplements of riboflavin may help maximize residual FMO3 enzyme activity15).In conclusion, we report the first Korean patient with TMAuria who had novel compound heterozygous mutation. TMAuria is no longer regarded as a rare disorder and specific investigation may be needed who has unpleasant odor like rotten fish. Through genetic test, we can distinguish affected individuals by the severe primary genetic form.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Ayesh R, Mitchell SC, Zhang A, Smith RL. The fish odour syndrome: biochemical, familial, and clinical aspects. BMJ 1993;307:655–657.
[Article] [PubMed] [PMC]2. van Thriel C, Schäper M, Kiesswetter E, Kleinbeck S, Juran S, Blaszkewicz M, et al. From chemosensory thresholds to whole body exposures-experimental approaches evaluating chemosensory effects of chemicals. Int Arch Occup Environ Health 2006;79:308–321.
[Article] [PubMed]3. Motika MS, Zhang J, Cashman JR. Flavin-containing monooxygenase 3 and human disease. Expert Opin Drug Metab Toxicol 2007;3:831–845.
[Article] [PubMed]4. Chalmers RA, Bain MD, Michelakakis H, Zschocke J, Iles RA. Diagnosis and management of trimethylaminuria (FMO3 deficiency) in children. J Inherit Metab Dis 2006;29:162–172.
[Article] [PubMed]5. Yamazaki H, Shimizu M. Survey of variants of human flavin-containing monooxygenase 3 (FMO3) and their drug oxidation activities. Biochem Pharmacol 2013;85:1588–1593.
[Article] [PubMed]6. Yi HG, Lee JN, Ryu SD, Kang JH, Cha YN, Park CS. Secondary fishodor syndrome can be acquired by nitric oxide-mediated impairment of flavin-containing monooxygenase in hepatitis B virus-infected patients. Korean J Physiol Pharmacol 2004;8:213–218.7. Cashman JR, Zhang J. Human flavin-containing monooxygenases. Annu Rev Pharmacol Toxicol 2006;46:65–100.
[Article] [PubMed]8. Hernandez D, Addou S, Lee D, Orengo C, Shephard EA, Phillips IR. Trimethylaminuria and a human FMO3 mutation database. Hum Mutat 2003;22:209–213.
[Article] [PubMed]9. Shimizu M, Allerston CK, Shephard EA, Yamazaki H, Phillips IR. Relationships between flavin-containing mono-oxygenase 3 (FMO3) genotype and trimethylaminuria phenotype in a Japanese population. Br J Clin Pharmacol 2014;77:839–851.
[Article] [PubMed] [PMC]10. Christodoulou J. Trimethylaminuria: an under-recognised and socially debilitating metabolic disorder. J Paediatr Child Health 2012;48:E153–E155.
[Article] [PubMed]11. Mountain H, Brisbane JM, Hooper AJ, Burnett JR, Goldblatt J. Trimethylaminuria (fish malodour syndrome): a “benign” genetic condition with major psychosocial sequelae. Med J Aust 2008;189:468
[Article]12. Mitchell SC, Smith RL. Trimethylaminuria: the fish malodor syndrome. Drug Metab Dispos 2001;29(4 Pt 2): 517–521.
[PubMed]13. Phillips IR, Shephard EA. Primary trimethylaminuria. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, , editors. GeneReviews(R). Seattle (WA): University of Washington, Seattle, 1993-2017.14. Messenger J, Clark S, Massick S, Bechtel M. A review of trimethylaminuria: (fish odor syndrome). J Clin Aesthet Dermatol 2013;6:45–48.15. Manning NJ, Allen EK, Kirk RJ, Sharrard MJ, Smith EJ. Riboflavin-responsive trimethylaminuria in a patient with homocystinuria on betaine therapy. JIMD Rep 2012;5:71–75.
[PubMed]