 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 61(5); 2018
Differentiation between incomplete Kawasaki disease and secondary hemophagocytic lymphohistiocytosis following Kawasaki disease using N-terminal pro-brain natriuretic peptide
-
Jung Eun Choi, MD1, Yujin Kwak, MD1, Jung Won Huh, MD2, Eun-Sun Yoo, MD1, Kyung-Ha Ryu, MD1, Sejung Sohn, MD1, Young Mi Hong, MD1

- Corresponding author: Young Mi Hong, MD. Department of Pediatrics, Ewha Womans University School of Medicine, 1071 Anyangcheon-ro, Yangcheon-gu, Seoul 07985, Korea. Tel: +82-2-2650-2841, Fax: +82-2-2653-3718, ymhong@ewha.ac.kr
- Received August 25, 2017 Revised March 13, 2018 Accepted March 17, 2018
- Abstract
-
- Purpose
- Purpose
- Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome with many causes, including Kawasaki disease (KD). The purpose of this study was to identify the laboratory tests needed to easily differentiate KD with HLH from incomplete KD alone.
- Methods
- Methods
- We performed a retrospective study on patients diagnosed with incomplete KD and incomplete KD with HLH (HLH-KD) between January 2012 and March 2015. We compared 8 secondary HLH patients who were first diagnosed with incomplete KD with all 247 incomplete KD diagnosed patients during the study period. The complete blood count, erythrocyte sedimentation rate, platelet count, and serum total protein, albumin, triglyceride, C-reactive protein, N-terminal pro-brain natriuretic peptide (NT-proBNP), and ferritin levels were compared. Clinical characteristics and echocardiography findings were also compared between the 2 groups.
- Results
- Results
- The total duration of fever was longer in the HLH-KD group than in the KD group. White blood cell and platelet counts were higher in the KD group. Alanine aminotransferase, ferritin, and coronary artery diameter were increased in the HLH-KD group compared with those in the KD group. The median of NT-proBNP was significantly higher in the HLH-KD group than in the KD group at 889.0 (interquartile range [IQR], 384.5–1792.0) pg/mL vs. 233.0 (IQR, 107.0–544.0) pg/mL.
- Conclusion
- Conclusion
- The NT-proBNP level may be helpful in distinguishing incomplete KD from KD with HLH. The NT-proBNP level should be determined in KD patients with prolonged fever, in addition to the white blood cell count, platelet count, and ferritin level, to evaluate secondary HLH.
- Introduction
- Introduction
Kawasaki disease (KD) is an acute febrile vasculitis in infants and young children, predominantly affecting medium-sized vessels, including coronary arteries. To date, there is no specific laboratory test to predict KD; hence, clinical criteria are used to make the diagnosis.1) The classical diagnostic criteria for KD by the American Heart Association (AHA) include the existence of high fever for more than 5 days and the presence of at least 4 of the 5 following criteria: (1) changes in extremities; (2) polymorphous exanthema; (3) bilateral bulbar conjunctival injection without exudates; (4) changes in lips and oral cavity; and (5) cervical lymphadenopathy (>1.5-cm diameter). Incomplete KD can also be diagnosed by the AHA guideline, including prolonged fever over 5 days and 2–3 of standard clinical features of KD.1)KD includes many associated complications such as cardiac, neurological, hematological, renal and gastrointestinal manifestations. Hematologic complications of KD include hemophagocytic syndrome (HLH)2) and macrophage activation syndrome (MAS).3)The characteristic feature of HLH appears to be an overgrowth of histiocytes with subsequent histiocytic phagocytosis found in the blood cells of the lymph nodes, bone marrow, liver and spleen. This probably represents a multi-system disorder characterized by a dysregulation of the immune system with hypercytokinemia and hyperinflammation.4)In previous studies, HLH was apparent in a prolonged or recurrent cases of KD.5,6,7,8,9,10,11,12,13) The mechanism by which KD develops into or triggers HLH has not been full elucidated. Abnormal immune-regulation of T cells resulting in hypercytokinemia has been proposed to contribute to the pathogenesis of both syndromes.4)A differential diagnosis between KD and HLH can be difficult due to the similarity of their symptoms and signs. Hypercytokinemia is believed to drive the overactivation of immune cells, resulting in tissue damage together with these cells.4,14,15)Current evidence suggests that N-terminal pro-brain natriuretic peptide (NT-proBNP) may be used as a diagnostic tool for KD. NT-proBNP has a high diagnostic value for identifying KD in patients with protracted undifferentiated febrile illness.16,17,18,19) There has not been a study on NT-proBNP in secondary HLH following KD. Hence, the purpose of this study was to compare the clinical and laboratory characteristics of incomplete KD and HLH-KD and to determine which biomarkers may be easily differentiate KD with HLH from incomplete KD alone.
- Materials and methods
- Materials and methods
- 1. Patients and materials
- 1. Patients and materials
The medical records of patients, who were diagnosed with incomplete KD and KD with HLH between January 2012 and March 2015, were retrospectively reviewed. The total of 255 children who met the diagnostic criteria for incomplete KD were enrolled. Incomplete KD can be diagnosed by the AHA guideline, including prolonged fever over 5 days and 2–3 of standard clinical features of KD.1) Eight secondary HLH patients who had previously been diagnosed with incomplete KD were compared with the 247 incomplete KD patients without HLH.2)The study was approved by the Institutional Review Board of Ewha Womans University Mokdong Hospital (approval number: 2017-11-017-002), and written informed consents were obtained from the parents of all subjects.- 2. Laboratory test
- 2. Laboratory test
We measured the following parameters at initial diagnosis of incomplete KD in both groups: white blood cell (WBC) count, percentage of neutrophil in WBC (% neutrophils), hemoglobin, platelet counts, aspartate aminotransferase (AST), alanine aminotransferase (ALT), erythrocyte sedimentation rate (ESR), serum total protein, albumin, triglyceride (TG), C-reactive protein (CRP) and ferritin. We measured the NT-proBNP at presentation of HLH in HLH-KD group. Moreover, comparisons of the basic patient characteristics were made, including laboratory values and echocardiographic findings between KD patients and HLH-KD patients.- 3. Diagnostic criteria
- 3. Diagnostic criteria
The diagnosis of incomplete KD by the AHA include prolonged unexplained fever, fewer than 4 of the principal clinical findings, and compatible laboratory or echocardiographic findings. Complete KD patients were excluded. We defined refractory KD patients as those who had recrudescent or persistent fever at least 36 hours after the end of initial intravenous immunoglobulin (IVIG) infusion.1)The diagnostic criteria of HLH is met when at least 5 of the 8 following criteria were fulfilled: (1) fever; (2) splenomegaly; (3) cytopenia affecting at least 2 of the 3 lineages in the peripheral blood ( hemoglobin <9 g/dL, in infants <4 weeks: hemoglobin <10 g/dL, platelets <100×103/mL, neutrophils <1×103/mL); (4) hypertriglyceridemia (fasting, 265 mg/dL) and/or hypofibrinogenemia (<150 mg/dL); (5) hemophagocytosis in bone marrow, spleen, lymph nodes, or liver; (6) low or absent natural killer-cell activity; (7) ferritin >500 ng/mL; (8) elevated sCD25 (soluble IL-2 receptor≥2,400/mL).2)- 4. Echocardiography
- 4. Echocardiography
Echocardiography was performed to detect the presence of coronary artery lesions by pediatric cardiologists using an IE33 machine (Philips Medical System, Andover, MA, USA) with a S8 and S5 transducer. Standard parasternal and apical views were taken. Two-dimensional echocardiography and M-mode echocardiogram, pulsed, color-flow Doppler and tissue Doppler imaging were also taken. Coronary arteries were categorized as abnormal if (1) the internal lumen diameter was >3 mm in children younger than 5 years of age and ≥4 mm in children aged ≥5 years; (2) the internal diameter of a segment measured ≥1.5 times that of an adjacent segment; or (3) the coronary lumen was clearly irregular.20)- 5. Bone marrow aspiration
- 5. Bone marrow aspiration
HLH is confirmed by bone marrow aspiration. The bone marrow finding shows histiocytic aggregation with hemophagocytic activity. The inserted photomicrographs shows hemophagocytosis, engulfed myelocytes and platelets (Wright stain, ×1,000) (Fig. 1).- 6. Treatment
- 6. Treatment
KD patients were treated with IVIG 2 g/kg for 10–12 hours. In IVIG-resistant patients with only KD, a second dose of IVIG 2 g/kg was administrated if fever lasted at least 36 hours after the end of IVIG infusion.1) In addition, methyl prednisolone (30 mg/kg for 3 consecutive days) or infliximab (5 mg/kg) were also applied to IVIG-resistant patients.For HLH-KD patients, the goal of therapy is to suppress life-threatening inflammation by destroying the immune cells.21,22) Therapy based on the HLH-2004 protocol consists of a series of weekly treatments with dexamethasone, cyclosporine and etoposide (VP-16). Intrathecal methotrexate and hydrocortisone are given to those with central nervous system disease. Some patients, especially familial type and refractory type, must also have their immune systems replaced by receiving a hematopoietic stem cell transplant in order to be cured of HLH.- 7. Statistical analysis
- 7. Statistical analysis
IBM SPSS Statistics ver. 24.0 (IBM Co., Armonk, NY, USA) was used for all the statistical analyses. As continuous data were not satisfied normality assumption via Shapiro-Wilk test, results were presented as median with interquartile range. In categorical data, the summary value was expressed as the number of subjects and percent. To compare the clinical characteristics and laboratory data of patients with HLH-KD and that of KD, numeric data were used for Mann-Whitney U test and categorical data were used for Fisher exact test. The correlation between NT-proBNP and other factors in HLH-KD patients were analyzed using Spearman correlation. A P<0.05 was considered statistically significant.
- Results
- Results
- 1. Clinical and laboratory findings between 2 groups
- 1. Clinical and laboratory findings between 2 groups
Table 1 shows the clinical characteristics between the incomplete KD group and the HLH-KD group. The median of age was younger in the KD group than the HLH-KD group 29.0 months (interquartile range [IQR], 12.0–48.0 months) vs. 62.0 months (IQR, 45.0–99.5 months), P=0.002. The total duration of fever was longer in the HLH-KD group than in the KD group 12.0 days (IQR, 11.0–14.5 days) vs. 5.0 days (IQR, 4.0–6.0 days), P<0.001 (Table 1). However, there were no significant differences in the sex, conjunctival injection, cervical lymphadenopathy, skin rash, abnormalities of lip or oral mucosa, abnormalities of extremities and BCGitis.According to the laboratory data, WBC 10.8×103/L (IQR, 8.4–14.1×103/L) vs. 5.3×103/L (IQR, 3.7–9.9×103/L), P=0.007 and platelet levels 292.0×109/L (IQR, 226.0–347.0×109/L) vs. 197.0×109/L (IQR, 144.5–236.0×109/L), P=0.013 were higher in the KD group compared with the HLH-KD group. Furthermore, AST 76.5 (IQR, 36.0–186.5) IU/L vs. 29.0 (IQR, 22.0–39.5) IU/L, P=0.004 was higher in the HLH-KD group compared with the KD group (Table 2).Ferritin was increased in the HLH-KD group compared with the KD group 3,122.0 ng/mL (IQR, 1,405.3–5,505.5 ng/mL) vs. 120.1 ng/mL (IQR, 77.0–171.0 ng/mL), P<0.001. Fig. 2 shows a scattergram of the high ferritin level in the HLH-KD patients. NT-proBNP were significantly higher in the HLH-KD group compared with the KD group 889.0 pg/mL (IQR, 384.5–17,92.0 pg/mL) vs. 233.0 pg/mL (IQR, 107.0–544.0 pg/mL), P=0.005 (Table 2).- 2. Echocardiography findings
- 2. Echocardiography findings
Echocardiography findings were also compared between the 2 groups. Coronary artery diameter was increased in the HLH-KD group compared with the KD group 2.1 mm (IQR, 1.7–3.5 mm) vs. 3.9 mm (IQR, 3.6–4.2 mm), P=0.012 (Table 2).- 3. Treatments
- 3. Treatments
At first, the 247 incomplete KD patients were treated with IVIG (2 g/kg/day) for 10–12 hours. Of these, 241 patients responded to primary IVIG infusion. The remaining 6 IVIG-resistant patients: 1 patient was treated with additional IVIG, 4 were treated with pulse intravenous methylprednisolone, and the other one was administrated infliximab. They were retreated if fever was observed at least 36 hours after the end of initial IVIG infusion (Fig. 3).In the case of HLH-KD patients, all 8 patients were initially treated with IVIG (2 g/kg/day). Second IVIG was administrated to four of eight IVIG-resistant patients, and infliximab was also applied to one of them (Table 3, Fig. 3).In the HLH-KD group, HLH-2004 chemo-immunotherapy was administrated. Mortality was observed in 2 out of 8 cases in the HLH-KD group. The average duration from KD to HLH onset was 7.5 days (range, 2–21 days). The interval between diagnosis and mortality was different from patient to patient; in one patient was 7 days, and the other case was 1 day (Table 3).- 4. The correlation analysis between NT-proBNP and other factors in HLH-KD patients
- 4. The correlation analysis between NT-proBNP and other factors in HLH-KD patients
NT-proBNP was significantly correlated with WBC (P=0.047). NT-proBNP was not correlated with other factors such as ESR, ferritin, TG, CRP, AST, platelet and coronary artery diameter (Table 4).
- Discussion
- Discussion
HLH is a clinical syndrome of hyperinflammation resulting in an uncontrolled and ineffective immune response with hypercytokinemia.18) Marked elevation of various cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-8, IL-10, IL-12, and IL-18, has been documented using either in animal models or clinical observation.4)Primary HLH is an autosomal recessive disorder caused by a number of different perforin signaling mutations.23) Secondary HLH can be triggered by infection, malignancy, drugs and immunological disease. As a serious complication of KD, secondary HLH is thought to result from a strong activation of the immune system, often caused by infection.24) Both KD and HLH have an inappropriate hypercytokinemia triggered by a variety of factors and greatly modified by genetic heterogeneity in response to infection or inflammation.25) To date, only primary HLH has been thought to involve genetic factors; however, recently, there is evidence of some degree of genetic predisposition even in secondary HLH.4)Early diagnosis and treatment of secondary HLH is important to prevent fatal outcomes. In our study, mortality was observed in 2 out of 8 cases in the HLH-KD group. Two mortality cases of HLH-KD were expired within 1 week after diagnosis of HLH. Of the 2 patients who treated with HLH-2004 protocol, one died of pneumonia and the other died of disease aggravation.Patients with secondary HLH treated with chemotherapy-based protocols have had only a 55% survival at the age of 3 years.23) Mortality in secondary HLH has been reported to vary from 8%–22% in rheumatologic HLH to 18%–24% in Epstein Bar virus HLH.26) An HLH series of 20 patients showed a mortality rate of 60%, and deaths were attributed to invasive infections in eight cases.27) In a previous Turkish pediatric study, Karapinar et al.28) reported that survival was 43% in critically ill pediatric patients whose HLH and multiple organ dysfunction syndrome (MODS) were treated with the HLH-94 2004 protocol. Recently published adult data showed an 89% mortality rate in patients with virus-associated HLH treated with chemoimmune therapy.29)Management of this syndrome relies on the early diagnosis, the identification of a triggering pathogen or an underlying disease, and management of lymphocyte/macrophage proliferation and activation. Severe cases are treated with chemotherapy, generally an etoposide-containing regimen, and hematopoietic stem cell transplantation is the treatment for familial, severe, and persistent nonfamilial cases.30)Patients with HLH frequently present prolonged high fever, progressive cytopenias, liver dysfunction, coagulopathy, and neurologic symptoms.13) In HLH-KD patient also had persistent fever with abnormal laboratory findings despite treatment of IVIG.There are various laboratory variables available for the diagnosis of HLH and the most widely used is ferritin. Hyperferritinemia has also been associated with HLH and many other inflammatory conditions such as sepsis, systemic inflammatory response syndrome, MODS, and MAS.14) The magnitude of elevation in serum ferritin has been associated with a worse prognosis.15) Our study showed an extremely elevated ferritin level in the HLH-KD group (Fig. 2).Recently, a number of studies have examined the predictive value of BNP or NT-proBNP for differentiating KD from other febrile illnesses. Serum BNP is elevated due to ventricular wall stress imposed by volume or pressure overload. In addition to the hemodynamic stress, inflammation of the myocardial tissue may also induce the production of BNP.23) It was also elevated in systemic inflammatory response. For clinical use, NT-proBNP may have some advantages over BNP. NT-proBNP has a longer half-life (60–120 minutes) than BNP (20–30 minutes) and the interpretation of serum levels of NT-proBNP is less affected by kidney functions. Another advantage of NT-proBNP is its stability at room temperature.17)In this study NT-proBNP was significantly higher in the HLH-KD group compared with the KD group. Furthermore, NT-proBNP was significantly correlated with WBC. NT-proBNP was not correlated with other factors such as ESR, ferritin, TG, CRP, AST, platelet, and coronary artery diameter. The highly elevated NT-proBNP is associated with cytokine reaction such as TNF-α and IL-1β.21) Therefore, the level of NT-proBNP can be increased by secondary HLH following KD with its mechanism originating from hypercytokinemia. Our data indicates that NT-proBNP is an important helpful diagnostic marker for the differentiation of incomplete KD from secondary HLH following KD.This study has some limitations to consider. There has not been enough study on level of NT-proBNP in secondary HLH. The small sample size in this study may weaken the statistical analytic power. Nationwide epidemiologic collaborative studies may be necessary to increase the volume of HLH. This was a retrospective chart review study, and diagnosis was drawn from clinical documentation.It should be noted that treatment for the underlying KD with IVIG, steroid, or infliximab had already been administered before the diagnosis of HLH, and thus may have influenced the clinical and laboratory findings.In conclusion, the laboratory tests to determine the levels of NT-proBNP may help to distinguish incomplete KD patients from KD with HLH patients.
- Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation 2017;135:e927–e999.
[Article] [PubMed]2. Kang HR, Kwon YH, Yoo ES, Ryu KH, Kim JY, Kim HS, et al. Clinical characteristics of hemophagocytic lymphohistiocytosis following Kawasaki disease: differentiation from recurrent Kawasaki disease. Blood Res 2013;48:254–257.
[Article] [PubMed] [PMC]3. Mukherjee D, Pal P, Kundu R, Niyogi P. Macrophage activation syndrome in Kawasaki disease. Indian Pediatr 2014;51:148–149.
[PubMed]4. Yang SL, Xu XJ, Tang YM, Song H, Xu WQ, Zhao FY, et al. Associations between inflammatory cytokines and organ damage in pediatric patients with hemophagocytic lymphohistiocytosis. Cytokine 2016;85:14–17.
[Article] [PubMed]5. Palazzi DL, McClain KL, Kaplan SL. Hemophagocytic syndrome after Kawasaki disease. Pediatr Infect Dis J 2003;22:663–666.
[Article] [PubMed]6. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001;85:421–426.
[Article] [PubMed] [PMC]7. Muise A, Tallett SE, Silverman ED. Are children with Kawasaki disease and prolonged fever at risk for macrophage activation syndrome? Pediatrics 2003;112(6 Pt 1): e495.
[Article] [PubMed]8. al-Eid W, al-Jefri A, Bahabri S, al-Mayouf S. Hemophagocytosis complicating Kawasaki disease. Pediatr Hematol Oncol 2000;17:323–329.
[Article] [PubMed]9. Kaneko K, Takahashi K, Fujiwara S, Maruyama T, Obinata K. Kawasaki disease followed by haemophagocytic syndrome. Eur J Pediatr 1998;157:610–611.
[Article] [PubMed]10. Ohga S, Ooshima A, Fukushige J, Ueda K. Histiocytic haemophagocytosis in a patient with Kawasaki disease: changes in the hypercytokinaemic state. Eur J Pediatr 1995;154:539–541.
[Article] [PubMed]11. Cummings C, McCarthy P, van Hoff J, Porter G Jr. Kawasaki disease associated with reactive hemophagocytic lymphohistiocytosis. Pediatr Infect Dis J 2008;27:1116–1118.
[Article] [PubMed]12. Titze U, Janka G, Schneider EM, Prall F, Haffner D, Classen CF. Hemophagocytic lymphohistiocytosis and Kawasaki disease: combined manifestation and differential diagnosis. Pediatr Blood Cancer 2009;53:493–495.
[Article] [PubMed]13. Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program 2009;127–131.
[PubMed]14. Demirkol D, Yildizdas D, Bayrakci B, Karapinar B, Kendirli T, Koroglu TF, et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care 2012;16:R52
[Article] [PubMed] [PMC]15. Sackett K, Cunderlik M, Sahni N, Killeen AA, Olson AP. Extreme Hyperferritinemia: causes and impact on diagnostic reasoning. Am J Clin Pathol 2016;145:646–650.
[Article] [PubMed]16. Dahdah N, Siles A, Fournier A, Cousineau J, Delvin E, Saint-Cyr C, et al. Natriuretic peptide as an adjunctive diagnostic test in the acute phase of Kawasaki disease. Pediatr Cardiol 2009;30:810–817.
[Article] [PubMed]17. Bae HK, Lee DK, Kwon JH, Kim HS, Sohn S, Hong YM. Clinical characteristics and serum N-terminal pro-brain natriuretic peptide as a diagnostic marker of Kawasaki disease in infants younger than 3 months of age. Korean J Pediatr 2014;57:357–362.
[Article] [PubMed] [PMC]18. Lin KH, Chang SS, Yu CW, Lin SC, Liu SC, Chao HY, et al. Usefulness of natriuretic peptide for the diagnosis of Kawasaki disease: a systematic review and meta-analysis. BMJ Open 2015;5:e006703.
[Article] [PubMed] [PMC]19. Kwon H, Lee JH, Jung JY, Kwak YH, Kim DK, Jung JH, et al. N-terminal pro-brain natriuretic peptide can be an adjunctive diagnostic marker of hyper-acute phase of Kawasaki disease. Eur J Pediatr 2016;175:1997–2003.
[Article] [PubMed]20. Japan Kawasaki Disease Research Committee. Report of subcommittee on standardization of diagnostic criteria and reporting of coronary artery lesions in Kawasaki disease. Tokyo: Ministry of Health and Welfare, 1984.21. Freeman HR, Ramanan AV. Review of haemophagocytic lymphohistiocytosis. Arch Dis Child 2011;96:688–693.
[Article] [PubMed]22. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–131.
[Article] [PubMed]23. Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol 2016;174:203–217.
[Article] [PubMed]24. Chen Y, Shang S, Zhang C, Liu T, Yang Z, Tang Y. Hemophagocytic lymphohistiocytosis at initiation of kawasaki disease and their differential diagnosis. Pediatr Hematol Oncol 2010;27:244–249.
[Article] [PubMed]25. Inoue S, Mangat C, Rafe'e Y, Sharman M. Forme Fruste of HLH (haemophagocytic lymphohistiocytosis): diagnostic and therapeutic challenges. BMJ Case Rep 2015;1 29 2015:pii: bcr2014206190
[Article]26. Kleynberg RL, Schiller GJ. Secondary hemophagocytic lymphohistiocytosis in adults: an update on diagnosis and therapy. Clin Adv Hematol Oncol 2012;10:726–732.
[PubMed]27. Sung L, King SM, Carcao M, Trebo M, Weitzman SS. Adverse outcomes in primary hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2002;24:550–554.
[Article] [PubMed]28. Karapinar B, Yilmaz D, Balkan C, Akin M, Ay Y, Kvakli K. An unusual cause of multiple organ dysfunction syndrome in the pediatric intensive care unit: hemophagocytic lymphohistiocytosis. Pediatr Crit Care Med 2009;10:285–290.
[Article] [PubMed]
Fig. 1
Bone marrow in a hemophagocytic lymphohistiocytosis-Kawasaki disease patient (5-year-old male) with hemophagocytic lymphohistiocytosis showed histiocytic aggregation with hemophagocytic activity. The photomicrograph shows hemophagocytosis, engulfed myelocytes, and platelets (Wright stain, ×1,000).
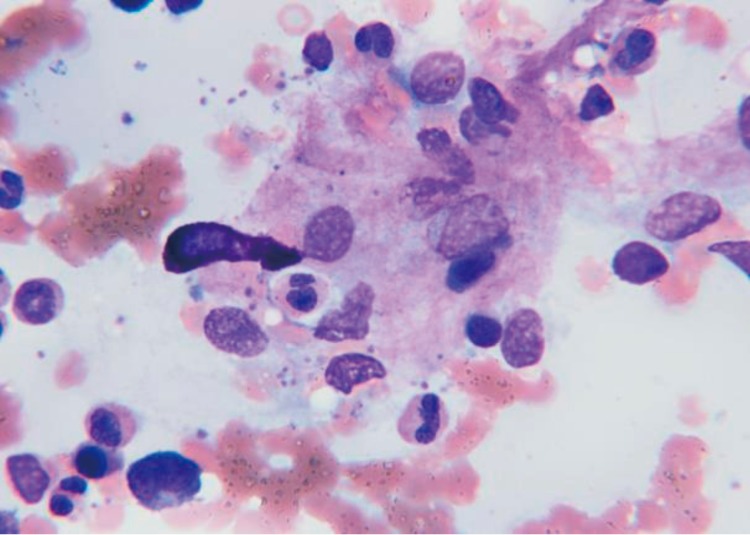
Fig. 2
Scattergram of ferritin levels in incomplete KD and HLH-KD patients. Ferritin level was significantly higher in HLH-KD patients than in incomplete KD patients. HLH, hemophagocytic lymphohistiocytosis; KD, Kawasaki disease.
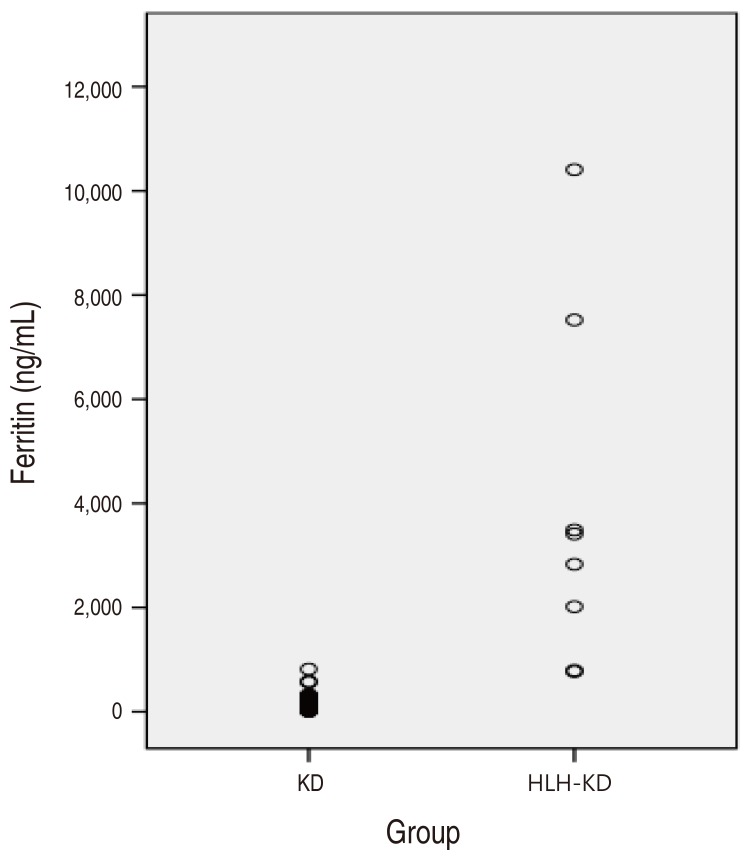
Fig. 3
Incomplete KD patients and medication in each group. HLH, hemophagocytic lymphohistiocytosis; KD, Kawasaki disease; IVIG, intravenous immunoglobulin; PD, prednisolone.

Table 1
Clinical characteristics of groups 1 and 2
Table 2
Laboratory data of groups 1 and 2
Values are presented as median (interquartile range).
Group 1, Kawasaki disease (KD); group 2, hemophagocytic lymphohistiocytosis-KD; Hb, hemoglobulin; WBC, white blood cell; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; AST, aspartate aminotransferase; ALT, alanine aminotransferase; NT-proBNP, N-terminal pro-brain natriuretic peptide.
Table 3
Treatment regimen and clinical course in HLH-KD patients