About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 59(Suppl 1); 2016
A novel mutation of CLCNKB in a Korean patient of mixed phenotype of Bartter-Gitelman syndrome
- Corresponding author: Heeyeon Cho, MD. Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel: +82-2-3410-3539, Fax: +82-2-3410-0043, choheeyeon@gmail.com
- Received November 25, 2015 Revised January 11, 2016 Accepted January 27, 2016
- Abstract
-
Bartter syndrome (BS) is an inherited renal tubular disorder characterized by low or normal blood pressure, hypokalemic metabolic alkalosis, and hyperreninemic hyperaldosteronism. Type III BS is caused by loss-of-function mutations in CLCNKB encoding basolateral ClC-Kb. The clinical phenotype of patients with CLCNKB mutations has been known to be highly variable, and cases that are difficult to categorize as type III BS or other hereditary tubulopathies, such as Gitelman syndrome, have been rarely reported. We report a case of a 10-year-old Korean boy with atypical clinical findings caused by a novel CLCNKB mutation. The boy showed intermittent muscle cramps with laboratory findings of hypokalemia, severe hypomagnesemia, and nephrocalcinosis. These findings were not fully compatible with those observed in cases of BS or Gitelman syndrome. The CLCNKB mutation analysis revealed a heterozygous c.139G>A transition in exon 13 [p.Gly(GGG)465Glu(GAG)]. This change is not a known mutation; however, the clinical findings and in silico prediction results indicated that it is the underlying cause of his presentation.
- Introduction
- Introduction
Bartter syndrome (BS), first described by Bartter and colleagues in 1962, is a group of rare autosomal recessive disorders, characterized by renal salt wasting, normal blood pressure, hypokalemic metabolic alkalosis with hyperreninemic hyperaldosteronism, and hyperplasia of the juxtaglomerular apparatus1). It is caused by impairment in sodium chloride reabsorption in the medullary thick ascending limb of the loop of Henle, and is characterized by its genetic and phenotypic heterogeneity2). BS is clinically categorized as antenatal BS and classic BS, and also categorized into 5 genetic subtypes. Underlying mutant genes for BS types I through V are the SLC12A1 gene encoding the sodium-potassium-chloride cotransporter (NKCC2) for type I, the KCNJ1 gene encoding the apical inwardly rectifying potassium channel (ROMK) for type II, the CLCKNB gene encoding the basolateral chloride channel (ClC-Kb) for type III, the BSND gene encoding the β-subunit for ClC-Ka and ClC-Kb for type IV, and the CASR gene encoding the basolateral calcium sensing receptor for type V3).In a nationwide cohort for Korean children with BS, CLCNKB mutations have been reported to be most common mutations, and there was no evidence of genotype-phenotype correlation3). In a cohort study, the Korean pediatric patients with CLCNKB mutations manifested from antenatal BS to mixed Bartter-Gitelman phenotypes3). In other populations, it has been also known that the clinical phenotype in patients with CLCNKB mutations can be highly variable, and clinical overlap of symptoms between BS and Gitelman syndrome (GS) is observed4,5). These studies suggested that a more clinically relevant classification in hereditary tubulopathies is necessary. We identified a novel missense mutation in CLCNKB in a Korean patient with atypical manifestations including severe chronic hypomagnesemia, hypokalemia, and hypercalciuria with nephrocalcinosis.
- Case report
- Case report
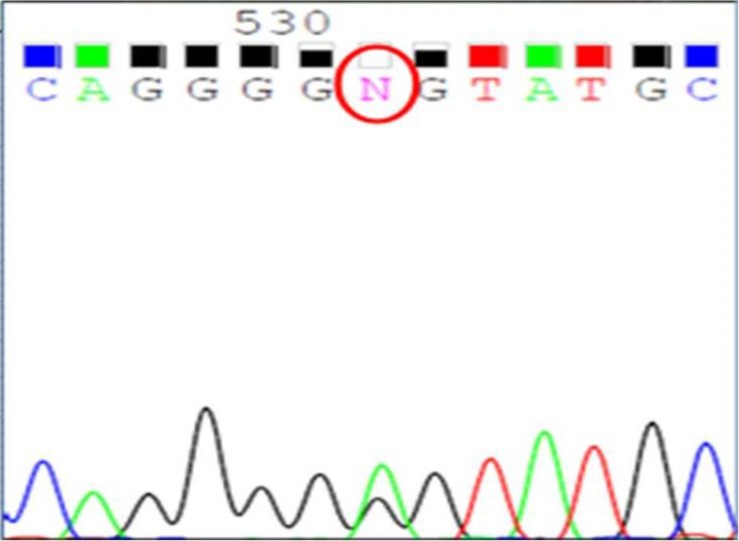
A 10-year-old male patient was referred to our hospital due to persistent hypokalemia and hypomagnesemia. He was born at 39+5 weeks gestational age by normal spontaneous vaginal delivery with a birth weight of 3,500 g. He had a family history significant for chronic hepatitis B in his mother and a cousin who underwent heart transplantation for unknown reasons. His birth history was significant for polyhydramnios. At the age of four years, he presented to an outside hospital with muscle cramps and rigidity. His height and weight were in the 50th percentile, and blood pressure was within normal range. Neurologic examination showed no abnormalities. However, laboratory and imaging findings showed severe hypomagnesemia, hypokalemia, hypocalcemia with hypercalciuria, elevated renin and aldosterone levels, and bilateral nephrocalcinosis. Renal wasting of magnesium and potassium was suspected, but an exact diagnosis was not made. Since then, he had been treated with oral potassium and magnesium supplements, amiloride, and spironolactone for 6 years. Despite oral and intermittent intravenous magnesium supplements, he remained hypomagnesemic. Genetic testing for familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) performed after 3 years of treatment was negative.At the age of 10 years, he was transferred to our hospital due to acute heart failure. He was diagnosed with acute myocarditis, and was managed with veno-arterial extracorporeal membrane oxygenation (VA-ECMO) for 13 days. Fortunately, his cardiac function recovered, though a definite etiology for the acute myocarditis was not identified. After successful weaning off of ECMO, he was transferred to the general ward and our pediatric nephrology department was consulted for evaluation of his chronic, persistent hypokalemia and hypomagnesemia.A thorough diagnostic evaluation was performed. His height was 145 cm (50th percentile) and body weight 36.5 kg (50th percentile). Serum sodium level was 132 mmol/L, potassium 3.2 mmol/L, and total CO2 20.2 mmol/L. Serum calcium level was 9.6 mg/dL, phosphorus 5.2 mg/dL, magnesium 1.1 mEq/L, blood urea nitrogen 6.7 mg/dL, creatinine 0.32 mg/dL. Urine specific gravity was 1.007 and there was no hematuria or proteinuria. In the spot urine sample, the calcium to creatinine ratio was 0.9. Tubular resorption of phosphate was 92.5% and the transtubular potassium gradient was 14.48 in spite of persistent hypokalemia. His clinical findings suggested a diagnosis of inherited renal tubulopathy, but his clinical manifestations were not straightforward, leading to confusion with regard to the subgroup diagnosis: BS type III, BS type V, GS or FHHNC were all suspected. After a thorough history taking, we discovered his antenatal history of polyhydramnios, which favored a diagnosis of BS type III. Ultimately, we analyzed the causative genes for BS type III, BS type V, GS and FHHNC (CLCNLKB, CASR, SLC12A3, and CLDN16, respectively). While the genetic studies for CLDN16, SLC12A3 and CASR were all negative, a heterozygous mutation in the CLCKNB gene was detected; c.139G>A causing p.Gly(GGG)465Glu(GAG) in exon 13 (Fig. 1). This is not a known mutation of the CLCKNB gene, and his clinical findings and in silico prediction suggested that this would very likely be pathogenic. The online programme “PolyPhen-2” classifies this amino acid exchange as probably damaging with a score 0.997 (sensitivity, 0.41; specificity, 0.98). After genetic confirmation of the diagnosis of BS type III, indomethacin therapy was started at a dosage of 0.05 mg/kg/day, and gradually increased up to 0.2 mg/kg/day without significant side effects. Oral magnesium and potassium supplements were maintained as well, with regular follow-up of laboratory tests and growth parameters.The present study was performed in accordance with the ethical standards of the institutional review board and with the Helsinki declaration.
- Discussion
- Discussion
According to the related defective genes, inherited renal tubular disorders associated with impaired renal conversion of salt, calcium, and magnesium are classified as BS, GS, isolated renal magnesium loss or FHHNC, and these disorders exhibit a broad spectrum of clinical pictures, often overlapping one another3). The genetic test is necessary to perform the correct diagnosis of the hereditary tubulopathies. Apart from BS, GS, and FHHNC were two other important hereditary tubulopathies included in the differential diagnosis in this case. FHHNC (OMIM*248250) is caused by mutations in the CLDN16 or CLDN19 gene encoding the tight junction proteins, claudin-16 and claudin-19, respectively6,7,8). It is characterized by massive hypermagnesuria and hypercalciuria leading to marked hypomagnesemia and nephrocalcinosis. A complete loss-of-function mutation of the CLDN16 gene is known to be associated with a more rapid decline in renal function9). Our patient had been initially suspected as having FHHNC, but after his referral to our hospital and being newly informed about the antenatal history of polyhydramnios, we suspected BS as the most likely diagnosis. While the antenatal history of polyhydramnios, early age of onset, hypercalciuria, and nephrocalcinosis were all more compatible with BS, our patient also manifested persistent hypomagnesemia, one of the typical characteristics that distinguishes GS from other tubulopathies. A number of previous studies point out that hypomagnesemia is present in almost all cases of GS, and is rare or absent in BS10,11). In our patient, severe hypomagnesemia was persistent in spite of the supplementation of magnesium and resulted in clinical symptoms. After the genetic diagnosis, oral indomethacin was given to the patient, and hypomagnesemia was improved with reduced dose of magnesium supplementation. This clinical finding is compatible with BS.BS type III (OMIM*607364), often known as classic BS, is caused by the dysfunction of ClC-Kb at the basolateral membrane of the thick ascending limb10). Among other types of inherited tubulopathies, it comprises the most heterogeneous clinical presentation, as compared to GS phenotypes11). There was a report of a novel missense mutation of CLCNKB in a Japanese patient of Gitelman-like phenotype with diuretic insensitiviry to thiazide administration4). A diuretic test using furosemide and thiazide as well as clinical findings has been used to make a differential diagnosis between BS and GS, but there were some reports that showed the discrepancies between the diuretic test and genotype4,5). It is recommended to perform the genetic test to diagnosis the hereditary tubulopathies. There are a few explanations for heterogeneity of BS. The heterogeneity of BS type III is probably attributed to the diversity in genetic defects and to the possible cooperation of ClC-Kb with other cotransporters and/or channels10,11). Andrini et al.10) described functional consequences of CLCKNB mutations and suggested that all ClC-Kb mutants show impaired membrane expression, and a minority of the mutants combine with a reduced expression and with an altered pH-dependent channel gating. The cooperation of ClC-Kb with other membrane proteins such as the ClC-Ka chloride channel or potassium-chloride cotransporter in the basolateral membrane of thick ascending limb epithelial cells has also been proposed as evidence for phenotypic variation10,12). There was a report a patient with BS type III caused by a novel mutation and single nucleotide polymorphisms (SNPs) in CLCNKB gene, and there is a possibility that a CLCNKB mutation combined with SNPs can cause the atypical clinical findings in BS13).Together, these findings suggest that the clinical distinction of inherited tubular disorders is not clear-cut and that the correlation between the underlying genetic mutation and the clinical phenotype is not universal. The phenotypic variation of hereditary tubular disorders has been consistently discussed over the past few decades, and there have been debates on how to clinically distinguish them from one another. Although it has been widely accepted that BS type III most commonly presents as classical BS, it may exhibit heterogeneous phenotypes overlapping with GS or FHHNC in a minority of patients, probably owing to variable functional consequences of mutations and the channel's possible interplay with other membrane cotransporters and channels10). Herein, we described our experience with such a case; a pediatric patient with atypical clinical findings caused by a novel CLCNKB gene mutation.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Bartter FC, Pronove P, Gill JR Jr, Maccardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med 1962;33:811–828.
[Article] [PubMed]2. Schurman SJ, Perlman SA, Sutphen R, Campos A, Garin EH, Cruz DN, et al. Genotype/phenotype observations in African Americans with Bartter syndrome. J Pediatr 2001;139:105–110.
[Article] [PubMed]3. Lee BH, Cho HY, Lee H, Han KH, Kang HG, Ha IS, et al. Genetic basis of Bartter syndrome in Korea. Nephrol Dial Transplant 2012;27:1516–1521.
[Article] [PubMed]4. Ohkubo K, Matsuzaki T, Yuki M, Yoshida R, Terawaki Y, Maeyama A, et al. A novel mutation of CLCNKB in a Japanese patient of Gitelman-like phenotype with diuretic insensitivity to thiazide administration. Meta Gene 2014;2:342–348.
[Article] [PubMed] [PMC]5. Nozu K, Iijima K, Kanda K, Nakanishi K, Yoshikawa N, Satomura K, et al. The pharmacological characteristics of molecular-based inherited salt-losing tubulopathies. J Clin Endocrinol Metab 2010;95:E511–E518.
[Article] [PubMed]6. Konrad M, Hou J, Weber S, Dotsch J, Kari JA, Seeman T, et al. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 2008;19:171–181.
[Article] [PubMed] [PMC]7. Praga M, Vara J, Gonzalez-Parra E, Andres A, Alamo C, Araque A, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Kidney Int 1995;47:1419–1425.
[Article] [PubMed]8. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 2006;79:949–957.
[Article] [PubMed] [PMC]9. Hanssen O, Castermans E, Bovy C, Weekers L, Erpicum P, Dubois B, et al. Two novel mutations of the CLDN16 gene cause familial hypomagnesaemia with hypercalciuria and nephrocalcinosis. Clin Kidney J 2014;7:282–285.
[Article] [PubMed] [PMC]10. Andrini O, Keck M, Briones R, Lourdel S, Vargas-Poussou R, Teulon J. ClC-K chloride channels: emerging pathophysiology of Bartter syndrome type 3. Am J Physiol Renal Physiol 2015;308:F1324–F1334.
[Article] [PubMed]11. Zelikovic I, Szargel R, Hawash A, Labay V, Hatib I, Cohen N, et al. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int 2003;63:24–32.
[Article] [PubMed]