 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 59(Suppl 1); 2016
Unusual malignant neoplasms of ovary in children: two cases report
- Corresponding author: Ali Ghribi, MD. Department of Pediatric Surgery, Children's Hospital of Tunis, Tunis El Manar University, Bab Saadoun 1007 Tunis, Tunisia. Tel: +216-7156810, Fax: +216-71562810, ali_ghribi_2007@yahoo.fr
- Received May 17, 2014 Revised October 13, 2014 Accepted November 07, 2014
- Abstract
-
Sex cord tumors with annular tubules are known to originate from the sex cord of embryonic gonads that synthesize Sertoli cells, Leydig cells, granulosa cells, and theca cells of the ovarian stroma, while ovarian small cell carcinoma of the hypercalcemic type is a type of neuroendocrine tumor. Both these tumors are uncommon, potentially malignant neoplasms in children. We report the case of a sex cord tumor with annular tubules in an 11-year-old girl and a case of small cell carcinoma of the hypercalcemic type in a 10-year-old girl. We also discuss the prognosis and management of these tumors.
- Introduction
- Introduction
Before puberty, the majority of ovarian neoplastic lesions are germ-cell tumors. Sex cord tumor with annular tubules (SCTAT) is an infrequent malignant ovarian tumor in children. SCTAT is a distinctive ovarian neoplasm. The predominant component of which has morphologic features intermediate between those of the granulosa cell tumor and those of the Sertoli cell tumor. Small cell carcinoma of the ovary of the hypercalcemia type (SCCOHT) is an uncommon and aggressive ovarian tumor that primarily affects young women and that is rarely observed in premenstrual adolescents. SCCOHT is a kind of neuroendocrine tumor. We report a case of SCTAT in an 11-year-old girl, and a case of a SCCOHT in a 10-year-old girl. The diagnosis of these 2 cases of unusual malignant neoplasms of ovary had not been suspected before surgery.
- Case reports
- Case reports
- 1. Case 1
- 1. Case 1
An 11-year-old girl was admitted to our department for intermittent pelvic pain that had been standing for a year as well as menarche followed by irregular episodes of uterine bleeding. The pain was exacerbated by walking and improved slightly with nonsteroidal anti-inflammatory medications. She was initially evaluated by her general pediatrician. Abdominal ultrasound examination revealed an intraabdominal mass measuring 20 cm in diameter. The mass was intraperitoneal and was found to consist of a cystic neoplasm. No shadowing was visualized to suggest calcification (Fig. 1). On examination, her breast development was Tanner stage 3 and pubic hair was Tanner stage 2. Clinical signs of Peutz-Jeghers syndrome (PJS) were absent. Ovarian tumor markers, including α-fetoprotein (α-FP) and human chorionic gonadotropin (HCG) were within normal range. At laparotomy, the right adnexa proved to be twisted but without necrosis. The tumor measuring 20 cm originated from the right ovary. It was a polycystic mass with a smooth surface filled with a clear yellowish fluid. The largest cyst measured 10 cm. The left ovary was grossly normal. No enlarged lymph nodes were found in the retroperitoneum. The omentum as well as the peritoneal surface were macroscopically unaffected. Tumor resection was carried out. Microscopic examination showed simple and complex ring-shaped tubules surrounding central hyaline material. The nuclei are located both at the periphery and centrally with intervening anuclear pale cytoplasm (Fig. 2A). Immunohistochemically, the tumor was strongly positive for calretinin (Fig. 2B), cytokeratin (Fig. 2C), vimentin and inhibin. Estradiol level in the cystic fluid was very high. A right salpingooophorectomy was performed 1 month later, because of the increased malignant potential of sex cord tumor with annular tubules. The patient had no event during 3 years of follow-up time.- 2. Case 2
- 2. Case 2
A 10-year-old girl was admitted to our department for acute abdominal pain. She had had an intermittent pelvic pain for 6 months without swelling or urinary frequency. There were no symptoms attributable to hypercalcemia. A history of weight loss could not be confirmed. On examination, the pelvis was tender without fever or any palpable mass. No features of precocious puberty were noted. Ultrasound examination showed a 6-cm solid right ovarian mass and ascites. There were no calcifications or lymphadenopathy (Fig. 3). An emergency laparotomy was performed revealing a necrotic right ovary due to torsion of an ovarian solid tumor. The left ovary looked normal and neither ascites nor adhesion was detected in the abdominal cavity. A right salpingo-oophorectomy was therefore performed. Histological and immunophenotypical studies concluded that it was a small-cell carcinoma of the ovary of the hypercalcemic type. Microscopy revealed prominent follicle-like spaces filled with eosinophilic fluid. Tumor cells were round and had scant cytoplasm (Fig. 4A). Immunohistochemistry is notable for pan-cytokeratin positivity and nuclear WT-1 staining. Pan-cytokeratin stain was positive (AE1/AE3/cytokeratin 7) (Fig. 4B, C). Subsequent laboratory tests revealed hypercalcemia (2.95 mmol/L). HCG, α-FP and cancer antigen 125 were normal. Chest X-ray, radionuclide bone scan and cerebral computed tomography scan were normal. The postoperative course was uneventful. In view of the highly malignant form of the tumor, the patient was given 6 courses of chemotherapy based on vinblastine (6 mg/m2 intravenous [IV] over 30 minutes on day 1), cisplatin (90 mg/m2 IV over 4 hours on day 1), and bleomycin (15 units/m2 IV over 24 hours on day 2). Seven years after the initial surgery, the patient was free of recurrent disease (normal calcium level, negative staging including chest radiography and abdominal ultrasound).
- Discussion
- Discussion
SCTAT represents 6% of sex cord stromal tumors and approximatively 5% of all ovarian neoplasms1,2). SCCOHT is a very rare tumor. A review of the literature notes only a few well-documented cases of SCCOHT in teenagers. SCTAT is known to origin from sex cord of embryonal gonads that make the sertoli cell leydig granulosa theca cell of the ovarian stroma, and the SCCOHT is a kind of neuroendocrine tumor1,3).The sole characteristic of SCTAT is based on histological finding along with the existence of simple and complex annular tubules1,4), like in our patient. Immunohistochemical analysis frequently reveals the expression of vimentin, cytokeratin and inhibin1). Tumor markers (HCG, α-FP) were negative for germ cell tumors. SCTAT was documented as an estrogen- and progesterone-secreting tumor5,6). Menometrorrhagia followed by persistent amenorrhea and a pelvic mass are important clinical features. There are 2 forms of tumors: SCTAT associated with PJS that manifests itself as typically bilateral, multifocal, with small cysts and SCTAT without PJS in which the tumors generally present as large and unilateral neoplasms2,5). In our first case report, the patient hadn't PJS but the mass was polycystic with large cysts that measured up to 10 cm. SCTAT associated with PJS is clinically benign. In women with non-PJS associated SCTATs, the malignant behavior with metastases may be seen in at least 20% of cases2,5). Given that, salpingo-oophorectomy may be recommended as primary surgery, even in young girls. The surgical procedure should include peritoneal washings, a thorough assessment of the pelvis and abdomen, omental biopsy, and ipsilateral pelvic and paraaortic lymph node sampling2). Tumorectomy should be followed by oophorectomy, in case of histological discovery of SCTAT, like in our patient. A recent retrospective study by Yinon et al.7) demonstrated that there was no statistically significant difference in tumor recurrence rates between patients who had undergone a cystectomy versus those who had undergone a unilateral salpingo-oophorectomy. In our case, considering the size of the tumor and its increased malignant potential and the patient's poor socio-economic conditions, we decided to perform a unilateral oophorectomy. Biopsy should be performed when the status of the contralateral ovary is in doubt6). If it is involved, cystectomy may be proposed as primary surgery to preserve fertility. In all cases, a strict follow-up is necessary. The serum tumor marker inhibin has been studied as predictor of the clinical behavior of the tumor postoperatively2). Long-term prognosis of this tumor in pediatric patients is unknown as there are sparse data in the literature2).SCCOHT is highly aggressive malignant tumor occurs mostly in young women3). The case studies of familial clustering of SCCOHT are suggestive of a heritable predisposition to SCCOHT, but the genetic etiology had yet to be characterized3). Histogenesis of SCCOHT and the mechanism of development of the hypercalcemia are unknown8,9). Histologically, the typical pattern is diffuse follicle-like sheets of small, closely packed cells with scant cytoplasm. The large cell variant consisting of the majority of cells with abundant cytoplasm is rare8). As in epithelial ovarian carcinomas, CA-125 could be a very useful marker3,10).The immunohistochemical staining pattern typical for tumor cells in SCCOHT is positive for epithelial markers (cytokeratin positive, epithelial membrane antigen), INI-1, WT-1, calretinin, CD10, and p53. Due to the often nonspecific morphology, other high grade neoplasms must be excluded, especially germ cell tumors, like dysgerminoma (negativity of HCG and α-FP). Our report had provided specific morphology and updated immunohistochemistry and the hypercalcemia had evoked the diagnosis. In fact, hypercalcaemia is present at the time of diagnosis in more than 60% of cases with SCCOHT3,9,10), like in our patient. The general symptoms are nonspecific and include an abdominal pain and a palpable mass. Symptoms like nausea and vomiting might be related to hypercalcemia. Severe hypercalcemia may cause serious sequelae, including hypotonia, lethargy, and coma and associated cardiac disturbances3,9,10). The combination of hypercalcemia and an ovarian mass in premenarchial girls should awaken the suspicion of SCCOHT. Radiological features are not specific as in sex cord tumor with annular tubules. According to the literature, prognosis of this tumor is extremely poor compared to other ovarian malignancies in children. Metastatic tumors have been shown to have a very diverse effect on survival3,4,5,6,7,8,9). Treatment approaches have included surgery usually followed by adjuvant chemotherapy, radiotherapy or both. The optimal surgical approach is unknown, but, as the disease is unilateral in 99% of cases, it seems unnecessary to perform a bilateral salpingooophorectomy and hysterectomy11). Unilateral oophorectomy associated with chemotherapy was widely applied. All long-term survivors, in the literature, received the combination of cisplatin and etoposide8,10). However, it is impossible to clearly distinguish the effects of chemotherapy and radiotherapy and to determine their individual influence on survival. Thus, the use of radiotherapy remains questionable11). Our patient received 6 courses of chemotherapy based on vinblastine, bleomycin and cisplatin without radiotherapy and with good outcome. Factors related to better outcome, in the literature, are age (>30 years), normal preoperative serum calcium, tumor size (<10 cm), and absence of large cells7,8,9,10).In conclusion, these 2 malignant potential tumors are unusual ovarian neoplasm subtype in children. Diagnosis may be referred by the clinic but often discovered by histology, like in our 2 case reports. The prognosis of these tumors is generally believed to be poor and complete surgical resection is strongly recommended, even in case of unsuspected diagnosis before surgery. Aggressive chemotherapy after surgery in case of SCCOHT seems to be mandatory. More reviews and analysis of case reports are important and represent the only way to gather information on very more tumor entities.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Schneider DT, Janig U, Calaminus G, Gobel U, Harms D. Ovarian sex cord-stromal tumors: a clinicopathological study of 72 cases from the Kiel Pediatric Tumor Registry. Virchows Arch 2003;443:549–560.
[Article] [PubMed]2. Nosov V, Park S, Rao J, Memarzadeh S. Non-Peutz-Jeghers syndrome associated ovarian sex cord tumor with annular tubules: a case report. Fertil Steril 2009;92:1497.e5–1497.e8.
[Article] [PubMed]3. McDonald JM, Karabakhtsian RG, Pierce HH, Iocono JA, Desimone CP, Bayliff SL, et al. Small cell carcinoma of the ovary of hypercalcemic type: a case report. J Pediatr Surg 2012;47:588–592.
[Article] [PubMed]4. Podczaski E, Kaminski PF, Pees RC, Singapuri K, Sorosky JI. Peutz-Jeghers syndrome with ovarian sex cord tumor with annular tubules and cervical adenoma malignum. Gynecol Oncol 1991;42:74–78.
[Article] [PubMed]5. Ishikawa H, Kiyokawa T, Takatani T, Wen WG, Shozu M. Giant multilocular sex cord tumor with annular tubules associated with precocious puberty. Am J Obstet Gynecol 2012;206:e14–e16.
[Article] [PubMed]6. Shen K, Wu PC, Lang JH, Huang RL, Tang MT, Lian LJ. Ovarian sex cord tumor with annular tubules: a report of six cases. Gynecol Oncol 1993;48:180–184.
[Article] [PubMed]7. Yinon Y, Beiner ME, Gotlieb WH, Korach Y, Perri T, Ben-Baruch G. Clinical outcome of cystectomy compared with unilateral salpingo-oophorectomy as fertility-sparing treatment of borderline ovarian tumors. Fertil Steril 2007;88:479–484.
[Article] [PubMed]8. Estel R, Hackethal A, Kalder M, Munstedt K. Small cell carcinoma of the ovary of the hypercalcaemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature. Arch Gynecol Obstet 2011;284:1277–1282.
[Article] [PubMed]9. Schleef J, Wagner A, Kleta R, Schaarschmidt K, Dockhorn-Dworniczak B, Willital G, et al. Small-cell carcinoma of the ovary of the hypercalcemic type in an 8-year-old girl. Pediatr Surg Int 1999;15:431–434.
[Article] [PubMed]
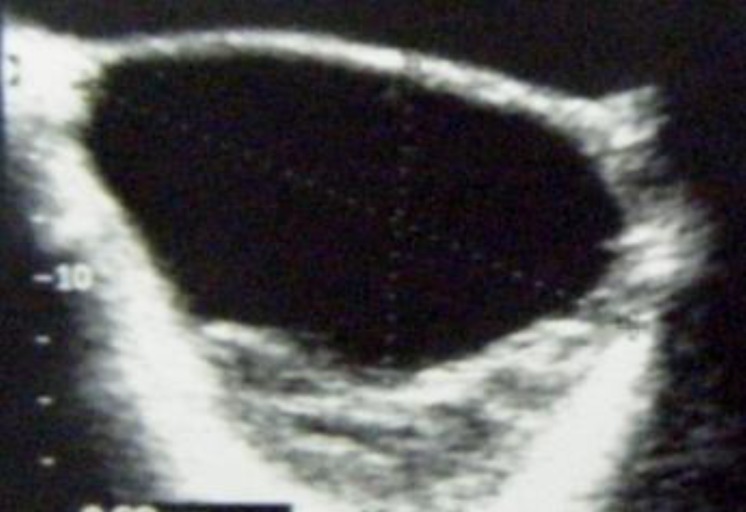
Fig. 2
(A) The tumor is composed of simple and complex ring-shaped tubules surrounding central hyaline material. The nuclei are located both at the periphery and in the center, with intervening anuclear pale cytoplasm (H&E, ×200). (B) The tumor cells show strong and diffuse positivity for calretinin (immunohistochemistry, ×200). (C) The tumor cells show diffuse positivity for cytokeratin (immunohistochemistry, ×200).
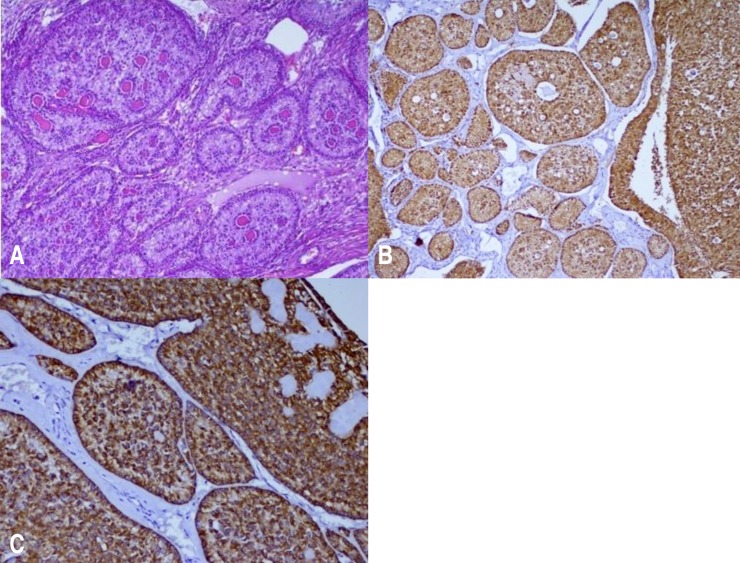
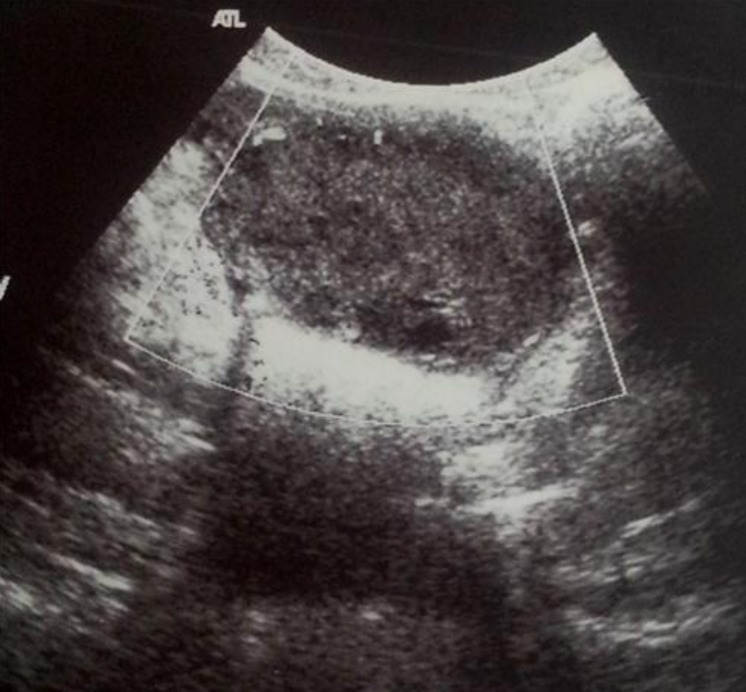
Fig. 4
(A) A primary ovarian small cell carcinoma of the hypercalcemic type. Prominent follicle-like spaces filled with eosinophilic fluid are present. The tumor cells are round and have scant cytoplasm (H&E, ×10). (B) The tumor cells show diffuse positivity for cytokeratin AE1–AE3 (immunohistochemistry, ×40). (C) The tumor cells show diffuse positivity for cytokeratin 7 (immunohistochemistry, ×40).
