 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 59(Suppl 1); 2016
1p36 deletion syndrome confirmed by fluorescence in situ hybridization and array-comparative genomic hybridization analysis
- Corresponding author: Jeesuk Yu, MD, PhD Department of Pediatrics, Dankook University Hospital, Dankook University College of Medicine, 201 Manghyang-ro, Dongnam-gu, Cheonan 31116, Korea. Tel: +82-41-550-6590, Fax: +82-41-565-6167, dryujs@dankook.ac.kr
- Received August 13, 2014 Revised August 13, 2014 Accepted October 23, 2014
- Abstract
-
Pediatric epilepsy can be caused by various conditions, including specific syndromes. 1p36 deletion syndrome is reported in 1 in 5,000–10,000 newborns, and its characteristic clinical features include developmental delay, mental retardation, hypotonia, congenital heart defects, seizure, and facial dysmorphism. However, detection of the terminal deletion in chromosome 1p by conventional G-banded karyotyping is difficult. Here we present a case of epilepsy with profound developmental delay and characteristic phenotypes. A 7-year- and 6-month-old boy experienced afebrile generalized seizure at the age of 5 years and 3 months. He had recurrent febrile seizures since 12 months of age and showed severe global developmental delay, remarkable hypotonia, short stature, and dysmorphic features such as microcephaly; small, low-set ears; dark, straight eyebrows; deep-set eyes; flat nasal bridge; midface hypoplasia; and a small, pointed chin. Previous diagnostic work-up, including conventional chromosomal analysis, revealed no definite causes. However, array-comparative genomic hybridization analysis revealed 1p36 deletion syndrome with a 9.15-Mb copy loss of the 1p36.33-1p36.22 region, and fluorescence in situ hybridization analysis (FISH) confirmed this diagnosis. This case highlights the need to consider detailed chromosomal study for patients with delayed development and epilepsy. Furthermore, 1p36 deletion syndrome should be considered for patients presenting seizure and moderate-to-severe developmental delay, particularly if the patient exhibits dysmorphic features, short stature, and hypotonia.
- Introduction
- Introduction
Pediatric epilepsy can be caused by many various conditions, including specific syndromes. It is useful to know the specific causes of epilepsy to understand its pathogenesis and for better management. Of all the known chromosomal abnormalities, the incidence of chromosome 1p36 alterations is not rare. The deletion of 1p36 occurs in 1 in 5,000–10,000 newborns, and terminal deletion is the most common deletion1,2,3,4). It also accounts for 0.5%–0.7% of the cases of idiopathic mental retardation5). This syndrome can be characterized by clinical features including developmental delay, mental retardation, hypotonia, congenital heart defects, seizure, and characteristic facial dysmorphism, but it is difficult to detect the terminal deletion of chromosome 1p by the conventional G-banded karyotypes4,6). With the microarray-based comparative genomic hybridization (array-CGH) analysis and fluorescence in situ hybridization (FISH) study with a subtelomeric 1p probe, the terminal 1p36 deletion can be confirmed1,6). Reported herein is a case of 1p36 deletion syndrome in a patient with recurrent febrile seizures and epilepsy, severe global developmental delay, hypotonia, congenital structural heart defects, and dysmorphic craniofacial features.
- Case report
- Case report
The 7-year- and 6-month-old boy was born from a 29-year-old mother. The family had no consanguinity and no specific remarkable family history of seizure, delayed development, hypotonia, or language delay. His 24-month-old younger brother was healthy.The patient was transferred from a local clinic for the evaluation of heart murmur at 9 days of life. At that time, a large patent ductus arteriosus (PDA) 4.2 mm in size, a small secundum atrial septal defect, and a small apical muscular ventricular septal defect were detected by Doppler echocardiography. The patient weighed 3.3 kg (less than 3rd percentile) and had a length of 53.1 cm (10th–25th percentile). During the follow-up, he still showed poor weight gain and a short stature, with microcephaly. At the age of 14 months, he weighed 7.4 kg (less than 3rd percentile) and had a length of 75.9 cm (10th–25th percentile) and a 43.8 cm head circumference (less than 3rd percentile). PDA ligation was done at the age of 4 months. According to the parents, the patient had a delayed gross motor milestone from head control. The Bayley study performed at the age of 6 and 24 months showed a significant delay in both the motor and mental aspects, respectively. The brain magnetic resonance imaging (MRI) that was done at the age of 5 months showed mild ventriculomegaly in the left lateral ventricle, and an arachnoid cyst (Fig. 1).When the patient was 5 years and 3 months old, he was admitted to the hospital due to afebrile generalized seizure. Before the admission, he had had recurrent febrile seizures since he was 12 months of age, with a frequency of 2 to 4 times per year, with a semiology of ocular deviation to the upward and generalized tonic-clonic type lasting for 30–60 seconds. During the patient's hospital stay, brain imaging and electroencephalography (EEG) were done (Figs. 1, 2). The interictal EEG showed moderate abnormality with intermittent bifrontal sharp waves, which were sometimes generalized (Fig. 2). As the baby's mother refused antiepileptic drugs medication, the natural course was observed without medication. At that time, the patient still showed severe global developmental delay, remarkable hypotonia, a short stature, and dysmorphic features such as microcephaly; small, low-set ears; dark, straight eyebrows; deep-set eyes; flat nasal bridge; midface hypoplasia; and a small, pointed chin (Table 1). The previous diagnostic work-up done at other hospitals, including the conventional chromosomal analysis, revealed no definite causes. As such, array-CGH analysis was done. The patient was rebrought to the hospital for the recurrence of seizure. An antiepileptic drug (valproic acid) was started at age 5 years and 11 months. The array-CGH analysis showed 1p36 deletion syndrome with a 9.15-Mb copy loss of the 1p36.33-1p36.22 region (Fig. 3). The FISH analysis confirmed the diagnosis (Fig. 4). At age 6 years and 10 months, the patient's developmental milestone showed 12 months of gross motor development, a 15-month fine motor status, an 18-month personal-social age, 18 months of language development, and a 16-month cognitive-adaptive status. He is now on a 120 mg valproic-acid twice a day (15 mg/kg/day) and has been seizure-free for 17 months.
- Discussion
- Discussion
It is well known that the deletion of the distal short arm of chromosome 1 (1p36 deletion) has typical clinical features, but it was considered that the prevalence of the syndrome may be underestimated due to the difficulty of detecting the light staining of the G-bands by trypsin using Giemsa-negative bands in the 1p36 region through the conventional chromosomal analysis4,6). With the recent advances in cytogenetic technology, especially array-CGH and FISH, it has become possible to identify the 1p36 deletions as well as the clinical phenotype and the molecular characteristics in detail4,7,8).Rosenfeld et al.7) described the common features of the monosomy 1p36 very clearly. The most common dysmorphic features of 1p36 deletion syndrome, which occur in more than 50% of the patients, are microcephaly, large anterior fontanelle, straight eyebrows, deep-set eyes, broad nasal root, midface hypoplasia, and pointed chin. The common neurologic manifestations included developmental delay, mental retardation, expressive language problems, neonatal hypotonia, seizures, eye/vision problems, hearing loss, and abnormal brain imaging7). The case reported herein also showed many characteristic clinical features, including congenital heart defects, severe global developmental delay, hypotonia, epilepsy, and characteristic craniofacial features. Due to the typical craniofacial features of 1p36 deletion syndrome, clinical suspicion is an important key to the diagnosis. It should be useful to perform a detailed chromosomal study such as array-CGH and/or FISH if the patient has significantly delayed development and epilepsy, especially with some of the characteristic morphologic features of monosomy 1p36.The prevalence of epilepsy was reported as 40%–73% in 1p36 deletion syndrome4,7,8,9,10). In most patients, seizure started in infancy, during the first 6 months of life, although it varied from neonate to 7 years. The seizure types were variable, and generalized tonic, tonic–clonic, or clonic types were common10,11). Infantile spasms were also reported11). The seizure outcome was usually favorable but could be severe and drug-resistant11). The case reported herein had recurrent febrile seizures since 12 months of age, and the first afebrile seizure occurred at the age of 5 years, but it was well controlled by valproic acid.The pathophysiological mechanisms of epilepsy in 1p36 deletion syndrome remain unclear. Recently, detailed chromosomal studies such as array-CGH and FISH led to a greater understanding of the phenotype of 1p36 deletion syndrome7). There are many well-defined genes in 1p36, including GNB1, GABRD, KCNAB2, PRKCZ, SKI, and PEX101,7,11). GNB1 (guanine nucleotide binding protein [G protein] beta polypeptide 1) encodes the β1 subunit of G protein. It regulates the alpha subunit of G protein and is likely to be involved in signal transduction in neurons12). GABRD (gamma-aminobutyric acid [GABA] A receptor, delta) encodes the subunit of the GABA-A receptors, and GABA is the major inhibitory neurotransmitter in the mammalian brain. Therefore, GABRD haploinsufficiency was suggested to play a role in the neurologic features, especially seizures7,13). The gene KCNAB2, a potassium voltage-gated channel from the shaker-related subfamily beta member 2, has been known as the first candidate gene attributed to seizures in monosomy 1p369,10), but someone without deletion of the KCNAB2 gene developed seizures, supporting the presence of other candidate genes (GABRD, PEX10) in seizures7,10,11). In this case, there are deletions of 3 genes: KCNAB2, GABRD, and PEX10. The gene PRKCZ (protein kinase C, zeta) encodes an atypical protein kinase C and is known to be necessary for mediating axonal differentiation and to be responsible for the memory7). The gene SKI (SKI proto-oncogene) is active in many cell types and plays a specific role in Schwann cell proliferation and myelination14). The haploinsufficiency of the SKI gene has been proposed as the possible cause of dysmorphic features, hypotonia, motor delay, mental retardation, and clefting in individuals with 1p36 deletion syndrome7,14,15).The case reported herein had an abnormality in the 1p36.33-1p36.22 region, with a 9.15 Mb copy loss, which contained the genes GNB1, CALML6, GABRD, KCNAB2, PRKCZ, SKI, and PEX10. As a consequence, it could be hypothesized that the haploinsufficiency of such genes may contribute to the patient's clinical phenotypes, such as profound global developmental delay, dysmorphic features, and epilepsy.On the matter of brain imaging, Bahi-Buisson et al.11) reported that cortical atrophy with enlargement of the lateral ventricles was the most common MRI feature of 1p36 deletion syndrome. The agenesis of the corpus callosum as a result of the copy number variations was also described in some literatures1,15). The haploinsufficiency of one gene in 1p36 deletions, SKI, was considered one of the causes of it7,15). The SKI gene is also known to play a role in myelination. Therefore, it could have been the cause of the demyelination shown in the patient's MRI. The patient also showed mild enlargement of the lateral ventricles as well as demyelination in the white matter along both corona radiata.In conclusion, it can be useful to consider a detailed chromosomal study in a case with delayed development and epilepsy. 1p36 deletion syndrome should be sought for a patient presenting seizure and moderate to severe developmental delay, particularly if the patient has dysmorphic features, a short stature, and hypotonia.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Gajecka M, Mackay KL, Shaffer LG. Monosomy 1p36 deletion syndrome. Am J Med Genet C Semin Med Genet 2007;145C:346–356.
[Article] [PubMed]2. Heilstedt HA, Ballif BC, Howard LA, Kashork CD, Shaffer LG. Population data suggest that deletions of 1p36 are a relatively common chromosome abnormality. Clin Genet 2003;64:310–316.
[Article] [PubMed]3. Shaffer LG, Lupski JR. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet 2000;34:297–329.
[Article] [PubMed]4. Shapira SK, McCaskill C, Northrup H, Spikes AS, Elder FF, Sutton VR, et al. Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am J Hum Genet 1997;61:642–650.
[Article] [PubMed] [PMC]5. Giraudeau F, Taine L, Biancalana V, Delobel B, Journel H, Missirian C, et al. Use of a set of highly polymorphic minisatellite probes for the identification of cryptic 1p36.3 deletions in a large collection of patients with idiopathic mental retardation. J Med Genet 2001;38:121–125.
[Article] [PubMed] [PMC]6. Riegel M, Castellan C, Balmer D, Brecevic L, Schinzel A. Terminal deletion, del(1)(p36.3), detected through screening for terminal deletions in patients with unclassified malformation syndromes. Am J Med Genet 1999;82:249–253.
[Article] [PubMed]7. Rosenfeld JA, Crolla JA, Tomkins S, Bader P, Morrow B, Gorski J, et al. Refinement of causative genes in monosomy 1p36 through clinical and molecular cytogenetic characterization of small interstitial deletions. Am J Med Genet A 2010;152A:1951–1959.
[Article] [PubMed]8. Heilstedt HA, Ballif BC, Howard LA, Lewis RA, Stal S, Kashork CD, et al. Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am J Hum Genet 2003;72:1200–1212.
[Article] [PubMed] [PMC]9. Heilstedt HA, Burgess DL, Anderson AE, Chedrawi A, Tharp B, Lee O, et al. Loss of the potassium channel beta-subunit gene, KCNAB2, is associated with epilepsy in patients with 1p36 deletion syndrome. Epilepsia 2001;42:1103–1111.
[Article] [PubMed]10. Kurosawa K, Kawame H, Okamoto N, Ochiai Y, Akatsuka A, Kobayashi M, et al. Epilepsy and neurological findings in 11 individuals with 1p36 deletion syndrome. Brain Dev 2005;27:378–382.
[Article] [PubMed]11. Bahi-Buisson N, Guttierrez-Delicado E, Soufflet C, Rio M, Daire VC, Lacombe D, et al. Spectrum of epilepsy in terminal 1p36 deletion syndrome. Epilepsia 2008;49:509–515.
[Article] [PubMed]12. Wang XB, Funada M, Imai Y, Revay RS, Ujike H, Vandenbergh DJ, et al. rGbeta1: a psychostimulant-regulated gene essential for establishing cocaine sensitization. J Neurosci 1997;17:5993–6000.
[Article] [PubMed] [PMC]13. Windpassinger C, Kroisel PM, Wagner K, Petek E. The human gamma-aminobutyric acid A receptor delta (GABRD) gene: molecular characterisation and tissue-specific expression. Gene 2002;292:25–31.
[Article] [PubMed]
Fig. 1
Brain magnetic resonance (MR) images of the patient at 5 months of age (A–C) and at 5 years and 3 months of age (D–F). The T2-weighted axial MR image at 5 months showed no abnormality in brain parenchyma with mild ventriculomegaly in left lateral ventricle, and myelination was compatible with 5 months of age (A). Follow-up image at 5 years and 3 months of age showed high signal intensities along both corona radiata (left>right) (arrow) (D). The T1-enhanced coronal MR image showed mild ventriculomegaly of left lateral ventricle (B, E), and T1-enhanced sagittal MR image showed arachnoid cyst or mega cisterna magna (C, F).
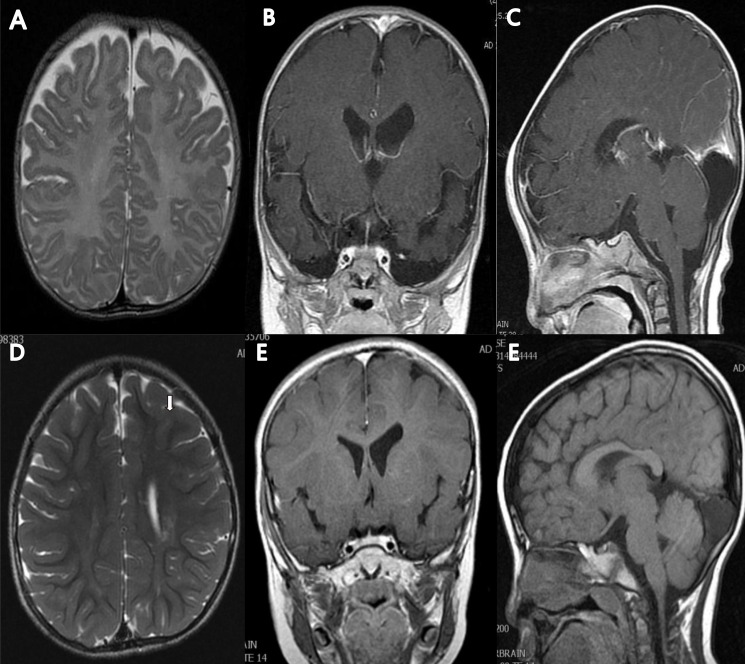
Fig. 2
Sleep electroencephalography showed moderately abnormal stage II sleep record due to intermittent bifrontal high amplitude sharp waves that are sometimes generalized.
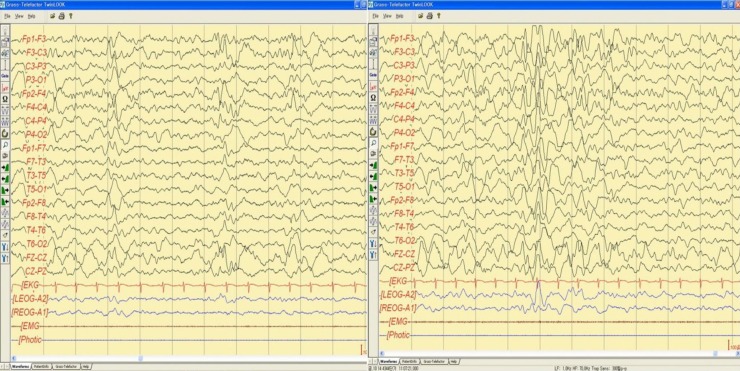
Fig. 3
Array-comparative genomic hybridization test revealed the site of deletion of 1p36 and the site of the breakpoint for the deletion of 1p36 (1p36.22-36.33).
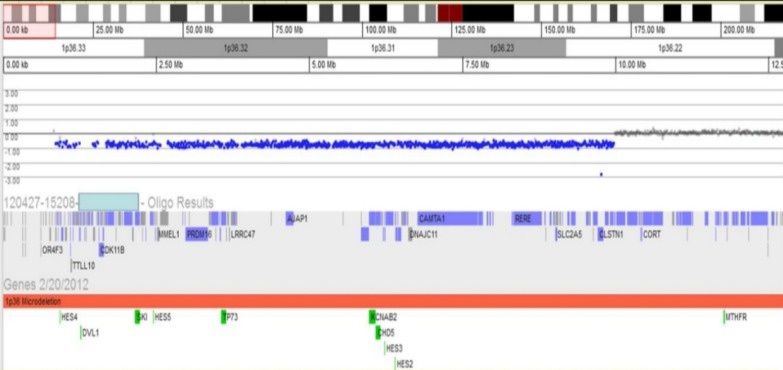
Fig. 4
Fluorescence in situ hybridization analysis using BAC RP11-425E15(1p36.33, spectrum red) and BAC RP 11-370K11(1q44, spectrum green) showed 46,XY,del(1p36.3).

Table 1
The clinical dysmorphic features and neurologic manifestations of the patient and their incidence in monosomy 1p36
*Based on the data from Rosenfeld et al.7)