 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 59(Suppl 1); 2016
Concurrency of Guillain-Barre syndrome and acute transverse myelitis: a case report and review of literature
- Corresponding author: Orkun Tolunay. Department of Pediatrics, Adana Numune Training and Research Hospital, Serinevler Mah. Ege Bagatur Blv. Adana, Turkey. Tel: +90-532-5948223, Fax: +90-322-3550101, orkuntolunay@yahoo.co.uk
- Received August 13, 2014 Revised January 03, 2015 Accepted January 27, 2015
- Abstract
-
Guillain-Barré syndrome and acute transverse myelitis manifest as demyelinating diseases of the peripheral and central nervous system. Concurrency of these two disorders is rarely documented in literature. A 4-year-old girl presenting with cough, fever, and an impaired walking ability was admitted to hospital. She had no previous complaints in her medical history. A physical examination revealed lack of muscle strength of the lower extremities and deep tendon reflexes. MRI could not be carried out due to technical problems; therefore, both Guillain-Barré syndrome and acute transverse myelitis were considered for the diagnosis. Intravenous immunoglobulin treatment was started as first line therapy. Because this treatment did not relieve the patient's symptoms, spinal MRI was carried out on the fourth day of admission and demyelinating areas were identified. Based on the new findings, the patient was diagnosed with acute transverse myelitis, and high dose intravenous methylprednisolone therapy was started. Electromyography findings were consistent with acute polyneuropathy affecting both motor and sensory fibers. Therefore, the patient was diagnosed with concurrency of Guillain-Barré syndrome and acute transverse myelitis. Interestingly, while concurrency of these 2 disorders is rare, this association has been demonstrated in various recent publications. Progress in diagnostic tests (magnetic resonance imaging and electrophysiological examination studies) has enabled clinicians to establish the right diagnosis. The possibility of concurrent Guillain-Barré syndrome and acute transverse myelitis should be considered if recovery takes longer than anticipated.
- Introduction
- Introduction
Guillain-Barre syndrome (GBS) is an acute inflammatory peripheral polyneuropathy consisting of 4 subtypes. Typically, acute inflammatory demyelinating polyradiculoneuropathy is observed with the initiation of an infection in the form of ascending symmetrical weakness and areflexia1,2). In aetiology it is often thought that as a result of infection based T-cell activation, antibodies are produced against myelin proteins of the peripheral nerves1).On the other hand, acute transverse myelitis (ATM) is a demyelinating disease with relatively sudden onset of motor, sensory and autonomic findings at the spinal cord3,4). One of the most common symptoms in children is pain (60%), followed by motor loss, weakness, numbness, ataxic walking, and loss of bladder or defecation control. Weakness mostly affects lower extremities. However, involvement of the upper extremities or even both extremities can also be observed. In the aetiology of both ATM and GBS, among other factors, autoimmunity triggered by infections is blamed.Although concurrency of demyelinating diseases of the central and peripheral nervous system are rare, in both disorders, autoimmunity triggered by infection conceives the possibility of this concurrency. However, this overlapping is rather rare and the causality is unknown.
- Case report
- Case report
A 4-year-old girl presenting cough, fever and incapability of walking, was admitted to hospital. The patient had a history of back pain and limping of the right foot for 5 days. The patient had no such complaints in the past. Physical examination revealed lack of muscle strength of the lower extremities (grade 2/5), upper extremities (grade 2/5) and the deep tendon reflexes. The patient did not suffer from neck stiffness. No meningeal irritation findings were observed either. Visual examination of the patient was normal. Sensory loss found in a transverse level of T9, with a noticeable decrease in sensation distally.Rales were detected during the examination of the respiratory system. Moreover, abdominal observation revealed urinary retention. An urethral catheter was inserted. Laboratory examination of the patient was as following: +++ leucocyte (before urethral catheterisation) was recorded in the urine examination; blood count, kidney functions, liver functions, and C-reactive protein revealed no abnormality; erythrocyte sedimentation rate was documented 20 mm/hr (normal range, 0–20 mm/hr); rheumatoid factor, antinuclear antibodies and antidouble stranded deoxyribonucleic acid antibodies were all negative. Complement levels (C3, C4) were normal. Vitamin B12 levels were normal. Serological studies for Mycoplasma pneumoniae, Toxoplasmosis, Rubella, Cytomegalovirus, Herpes simplex virus, Ebstein-Barr virus, Brucella, Salmonella, Listeria monocytogenes and hepatitis were negative.Bilateral interstitial opacities were detected in the chest X-ray. Urine culture result was positive for Escherichia coli (>103 colony forming unit/mL). A lumbar puncture was performed; the cerebrospinal fluid revealed normal protein and glucose levels with no pleocytosis. The cerebrospinal fluid culture yielded no growth of bacteria.Due to technical problems, magnetic resonance imaging (MRI) of the spine could not be done. Both GBS and also ATM were considered for the diagnosis. Intravenous immunoglobulin (IVIG) treatment (400 mg/kg/day, for 5 days) was started as first line treatment. Intravenous ceftriaxone and clarithromycin were administered following the initial diagnosis of urinary tract infection and bronchopneumonia.The MRI of the brain revealed no abnormality. A spinal MRI was carried out on the fourth day of the IVIG treatment. The spinal MRI exhibited high contrast retention medullary lesions on T2A with scattered and skipped settlement at the C7 - Th12 levels. After an intravenous contrast substance injection was made, the same MRI also revealed isointense signalled and patched contrast retention medullary lesions at T1A (Fig. 1). There was no sign of spinal cord compression, ischemia, tumour, or arteriovenous malformation on the MRI.There was no clinical response on the fourth day of the IVIG treatment. After the confirmation of ATM of the spinal MRI, pulse methylprednisolone (30 mg/kg/day) was administered for three days followed by oral prednisolone (1 mg/kg/day) for 1 month.In the electrophysiological study, no F wave responses could be obtained in any of the motor nerves of upper and lower extremities. Moreover, motor-sensory response amplitudes were lowered and finally, no sensory response could be received either. The electromyography findings were consistent with acute polyneuropathy affecting both motor and sensory fibers (Table 1). So, the patient was diagnosed with concurrency of GBS and ATM.On the 13th day of the patient's hospitalization, muscle strength was noted to be improved (grade 4/5), sensorial examination was normal and deep tendon reflexes were normalized. The patient was followed up after discharge. Within the first month of postdischarge follow-up, muscle strength was grade 4/5 during physical examination and deep tendon reflexes and sensorial examination were evaluated to be normal again.
- Discussion
- Discussion
GBS and ATM are basically autoimmune diseases. Because of an unknown cause in GBS, it is always thought that antibodies are produced against the myelin proteins of peripheral nerves resulting from T-cell activation1). Studies point out the cross reaction between the infection induced antibodies and the neuronal antigens, resulting in inflammatory neuropathies.All forms of GBS including Miller Fisher syndrome can be seen after a Campylobacter jejuni or a haemophilic influenza infection. The reason of that are similar structures like GM1 and GQ1b proteins existing at the haemophilic influenza wall5). GBS is also seen after Cytomegalovirus infections. It's been shown that GM2 based antibodies were evolved after Cytomegalovirus infections6). Five percent of GBS cases are preceded by M. pneumonia infections. There are only two cases documented in literature with concurrent GBS and E. coli7).In the aetiology of both ATM and GBS, infections are blamed. Infection triggered antibodies harm myelin structures on the medulla spinalis3). Suspected microorganisms of this occurrence are influenza virus, varicella, measles, mumps, dengue virus, and Mycoplasma. However, the correlation of ATM and E. coli has not been documented in literature yet.Despite urinary culture being positive of E. coli; initial clinical symptoms of lower respiratory infection and urinary retention revealed a secondary urinary infection. E. coli and its neurologic complications mostly evolve with strains (O157:H7 serotype) producing shiga toxin. The relation between E. coli and demyelinating diseases has been first documented by Kono et al.7) in 2007 on a patient with GBS and urosepsis.The aetiologies of both GBS and ATM comprise infection triggered autoimmunity. To test the theory, ganglioside like lipopolysaccharides, obtained from Campylobacter jejuni bacterial wall were injected to rabbits. Thereby, an outcome similar to acute motor axonal neuropathy was obtained8). Kono et al.7) detected antibodies against GM1 and GalNAc-GD1a but could not assess the relationship between the liposaccharides in the E. coli capsule and the antibodies.Although concurrency of GBS and ATM is rare, transitions between demyelinating diseases like GBS and ATM are possible. For the first time in 2001, a case of ATM and GBS occurring after a mumps virus infection was published by Bajaj et al.9) Until now, 7 concurrencies of GBS and ATM have been documented in literature. Six of these cases were published in 2007 and afterwards10,11,12,13,14,15). As manifested in literature, the responsible organisms in aetiology of concurrent GBS and ATM are Mycoplasma, influenza, Bartonella henselae and mumps (Table 2).We think that the overlapping of these 2 diseases is not a new case. But then, the question arises how this concurrency could be ignored all these years and how could the patients be treated without a proper diagnosis? IVIG is the first choice of therapy at GBS. Plasmapheresis is the second choice in case of a failure of IVIG2). If initial corticosteroid and plasmapheresis therapies do not work in severe cases with ATM, other therapy options such as IVIG or cyclophosphamide can also be taken into consideration3).In the GBS, no benefit was recorded by using corticosteroids alone2). In the light of this knowledge, if therapies other than solely corticosteroids are applied, patients might benefit from the treatment despite shortcomings in diagnosis.Using corticosteroids alone is not a choice in treatment of patients with concurrency of GBS and ATM. Studies reveal that IVIG was administered to 2 patients, IVIG+corticosteroids to 3 patients and plasmapheresis to 2 patients (Table 2). To accentuate, therapy modalities of the 7 cases documented in literature also support our thesis.It is also substantial to reiterate that the similarity in both of the mechanisms of the 2 diseases and also the treatment choices increase the possibility of a misdiagnosis of these diseases.It is not the first time within the last 10 years that concurrency of GBS and ATM occurred. Advances in diagnostic tests and also easier access to these tests have a major role in facilitating diagnoses.It should also not be forgotten that the two demyelinating diseases can be observed together in patients with GBS or ATM who do not respond to therapies or whose recovery takes more time than anticipated.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 2. Vucic S, Kiernan MC, Cornblath DR. Guillain-Barre syndrome: an update. J Clin Neurosci 2009;16:733–741.
[Article] [PubMed]3. Wolf VL, Lupo PJ, Lotze TE. Pediatric acute transverse myelitis overview and differential diagnosis. J Child Neurol 2012;27:1426–1436.
[Article] [PubMed]4. Berger JR, Cambi F, Di Rocco A, Farace J. Overview to approach to the patient with noncompressive myelopathy. Continuum (Minneap Minn) 2005;11:13–24.
[Article]5. Koga M, Yuki N, Tai T, Hirata K. Miller Fisher syndrome and Haemophilus influenzae infection. Neurology 2001;57:686–691.
[Article] [PubMed]6. Susuki K, Odaka M, Mori M, Hirata K, Yuki N. Acute motor axonal neuropathy after Mycoplasma infection: evidence of molecular mimicry. Neurology 2004;62:949–956.
[Article] [PubMed]7. Kono Y, Nishitarumizu K, Higashi T, Funakoshi K, Odaka M. Rapidly progressive Guillain-Barré syndrome following Escherichia coli infection. Intern Med 2007;46:589–591.
[Article] [PubMed]8. Yuki N, Susuki K, Koga M, Nishimoto Y, Odaka M, Hirata K, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barre syndrome. Proc Natl Acad Sci U S A 2004;101:11404–11409.
[Article] [PubMed] [PMC]9. Bajaj NP, Rose P, Clifford-Jones R, Hughes PJ. Acute transverse myelitis and Guillain-Barré overlap syndrome with serological evidence for mumps viraemia. Acta Neurol Scand 2001;104:239–242.
[Article] [PubMed]10. Bonastre-Blanco E, Jordan-Garcia Y, Fons-Estupina MC, Medina-Cantillo J, Palomeque-Rico A. Plasmapheresis in a paediatric patient with transverse myelitis and Guillain-Barre syndrome secondary to infection by Mycoplasma pneumoniae. Rev Neurol 2011;53:443–444.
[PubMed]11. Carman KB, Yimenicioglu S, Ekici A, Yakut A, Dinleyici EC. Co-existence of acute transverse myelitis and Guillain-Barré syndrome associated with Bartonella henselae infection. Paediatr Int Child Health 2013;33:190–192.
[Article] [PubMed]12. Howell KB, Wanigasinghe J, Leventer RJ, Ryan MM. Concomitant transverse myelitis and acute motor axonal neuropathy in an adolescent. Pediatr Neurol 2007;37:378–381.
[Article] [PubMed]13. Kang SH, Yum MS, Lee EH, Ko TS. Concurrent gullain-barre syndrome and acute transverse myelitis as an initial presentation of systemic lupus erythematosus. J Korean Child Neurol Soc 2012;20:121–128.
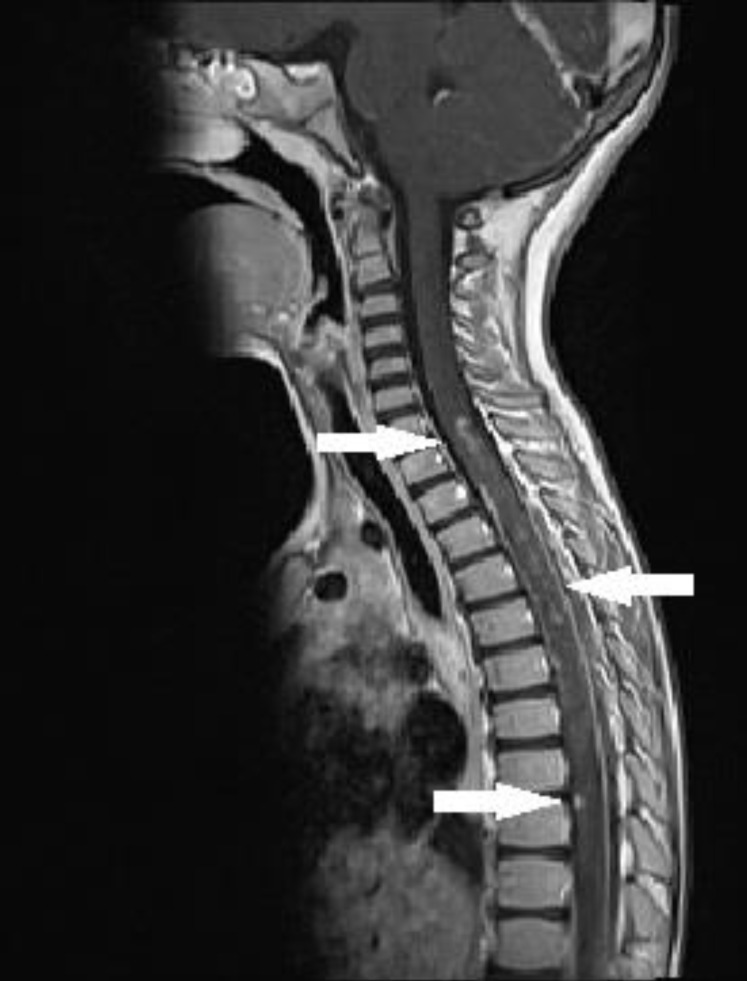
Table 1
Electromyographic findings from our patient
Table 2
Concurrency of GBS and TM in the literature
| Source | Year | Etiology | Treatment |
|---|---|---|---|
| Bajaj et al.9) | 2001 | Mumps | IVIG |
| Howell et al.12) | 2007 | ? | IVIG+corticosteroid |
| Tripp15) | 2008 | Influenza | Plasmapheresis |
| Bonastre-Blanco et al.10) | 2011 | M. pneumoniae | Plasmapheresis |
| Kang et al.13) | 2012 | Autoimmun | IVIG+corticosteroid |
| Topcu et al.14) | 2013 | M. pneumoniae | IVIG |
| Carman et al.11) | 2013 | Bartonella henselae | IVIG+corticosteroid |