 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 59(Suppl 1); 2016
A neonate with Joubert syndrome presenting with symptoms of Horner syndrome
- Corresponding author: Yun-Jin Lee, MD, PhD. Department of Pediatrics, Pusan National University Yangsan Hospital, 20 Geumo-ro, Mulgeum-eup, Yangsan 50612, Korea. Tel: +82-55-360-2180, Fax: +82-55-360-2181, jinnyeye@hanmail.net
- Received January 12, 2015 Revised April 08, 2015 Accepted May 21, 2015
- Abstract
-
Joubert syndrome (JS) is characterized by the “molar tooth sign” (MTS) with cerebellar vermis agenesis, episodic hyperpnea, abnormal eye movements, and hypotonia. Ocular and oculomotor abnormalities have been observed; however, Horner syndrome (HS) has not been documented in children with JS. We present the case of a 2-month-old boy having ocular abnormalities with bilateral nystagmus, left-dominant bilateral ptosis, and unilateral miosis and enophthalmos of the left eye, which were compatible with HS. Brain magnetic resonance imaging (MRI) revealed the presence of the MTS. Neck MRI showed no definite lesion or mass around the cervical sympathetic chain. His global development was delayed. He underwent ophthalmologic surgery, and showed some improvement in his ptosis. To the best of our knowledge, the association of HS with JS has not yet been described. We suggest that early neuroimaging should be considered for neonates or young infants with diverse eye abnormalities to evaluate the underlying etiology.
- Introduction
- Introduction
Joubert syndrome (JS) was initially described in 1968 in 4 siblings with agenesis of the cerebellar vermis presented with episodic hyperpnea, abnormal eye movements, ataxia, and intellectual disability1). Several years later, a pathognomonic midbrain-hindbrain malformation—the “molar tooth sign” (MTS)—was detected first in JS2), and then in several other conditions previously considered as distinct entities. This neuroradiological sign is now represented as a mandatory criterion for the diagnose of JS.JS can be assumed in all infants presented with hypotonia, altered respiratory pattern, abnormal eye movement, and developmental delay3). In the newborn period, episodic hyperpnea, a subtle facial appearance, and abnormal eye movements are helpful in making a diagnosis4). Nystagmus, colobomas, strabismus, ptosis, and retinal pigmentation are associated with eye abnormalities in neonates3). Concomitant symptoms of ptosis, anisocoria with unilateral miosis, and unilateral enophthalmos, which are compatible with Horner syndrome (HS), have not been documented in the children of JS children thus far.We now describe a neonate of JS presented with hypotonia and unique eye abnormalities of nystagmus, ptosis, anisocoria, and unilateral enophthalmos. To the best of our knowledge, JS with symptoms of HS has not been reported until now.
- Case report
- Case report
A 2-month-old boy was referred to the pediatric neurology clinic due to bilateral nystagmus, bilateral ptosis with left-dominant, left-sided enophthalmos, and anisocoria since the second week after birth. The patient was the first baby of a healthy 28-year-old mother and a healthy 32-year-old father. The baby was a 3,050 g (10th–25th percentile) infant born at gestational age of 38 weeks by normal vaginal delivery after an uneventful pregnancy. His height was 48.5 cm (25th–50th percentile), with a head circumference of 33 cm (10th–25th percentile). After birth, the baby had laryngomalacia, hypotonia, and a systolic murmur best heard at the upper left sternal border. An electrocardiogram and echocardiogram revealed normal sinus rhythm, and a small sized secundum atrial septal defect. No history of trauma and operation was noted.On physical examination, both pupils promptly reacted to light, but unilateral miosis and enophthalmos of the left eye and left-dominant bilateral ptosis were observed, which could be indicative of HS (Fig. 1A). Furthermore, strabismus was observed. He had a broad nasal bridge, and a triangular-shaped open mouth. He was hypotonic, and had intact deep tendon reflexes. Dysphagia and respiratory disturbance such as hyperpnea or apnea were not noted. Other physical examination findings were unremarkable. In order to evaluate the symptoms of HS and horizontal nystagmus, magnetic resonance imaging (MRI) of the orbit, brain and neck were performed at 2 to 3 months of age. Orbit and brain MRI showed hypoplastic cerebellar vermis and a narrowed anteroposterior diameter of the midbrain with a characteristic of ‘molar tooth appearance’ (Fig. 2), which were compatible with typical findings of JS. Neck MRI showed no definite lesion or mass around the cervical sympathetic nerve chain, thymus, and mediastinum as the underlying cause of HS. Laboratory examinations including renal and hepatic function tests and an abdominal ultrasonography showed normal findings. A chromosomal study revealed a normal 46XY. Left-sided visual loss was found in ophthalmological examinations, and neither coloboma nor pigmentary retinal changes were observed. No abnormality was noted in an audiological test and follow-up electrocardiogram and echocardiogram. Molecular genetic test was not performed.Clinical development at the age of 10 months was global delayed, as explained by the following: he could not babble, follow with or turn his eyes on objects, control his head, sit independently, or hold his bottle. He underwent ophthalmologic surgery (bilateral frontalis operation) due to persistent visual disturbance by bilateral ptosis, and had distinct improvement of ptosis; however, unilateral enophthalmos, and anisocoria remained (Fig. 1B). He is currently actively undergoing rehabilitation therapy, and will be regularly monitored through comprehensive developmental examination and follow-up imaging tests, including hepatorenal, cardiac, and ophthalmologic test.The gross photos of the figure were permitted by his parents through the informed consent.
- Discussion
- Discussion
JS is a rare autosomal-recessive disorder, which is characterized by midbrain-hindbrain malformation mainly in the form of agenesis or dysgenesis of cerebellar vermis3). The incidence rate is 1 out of 800,000 to 100,000 per a year in the newborn5). Incecik et al.5). describe several diagnostic criteria; (1) cranial MRI findings demonstrating the MTS on axial imaging with these three components: midline cerebellar vermis hypoplasia, deepened interpeduncular fossa, and thick, elongated superior cerebellar peduncles, (2) hypotonia in infancy, (3) global developmental delay/mental retardation, and (4) one or both of the following (not absolutely required but supportive of the diagnosis): (i) irregular breathing pattern in infancy (episodic tachypnea and/or apnea), and (ii) abnormal eye movement (nystagmus, jerky eye movements, and difficulty with smooth visual pursuits).Pathological findings of MTS correspond to an image of severe hypo-dysplasia of the cerebellar vermis with midline clefting, fragmentation of cerebellar nuclei and heterotopias of Purkinje-like neurons, along with dysplasia of the pontine and medullary structures such as the basis pontis, reticular formation, inferior olivary, dorsal column, and solitary tract nuclei. Moreover, typical findings are expressed by the lack of decussation both of the superior cerebellar peduncles and of the corticospinal tracts at the medullary pyramids6).A diagnosis of JS should be assumed in all infants presented with hypotonia, abnormal eye movement and developmental delay. Especially, a neonate of JS shows the characteristic physical signs and symptoms (Table 1)3). The infant may lie in a froglike position, and moderate to severe hypotonia can be observed3). The most common presented feature in the newborn period is an abnormal breathing pattern, characterized by episodic hyperpnea2,3). The distinctive short alternate episodes of apnea and hyperpnea or episodic hyperpnea lone, sometimes described as ‘panting like a dog,’ may intensify with stress and progressively improve with age, usually disappearing around sixth months2,3). Our patient had hypotonic muscle tone, but no respiratory abnormality.We found and briefly reviewed the reported patients of JS in Korea. The 7 children were diagnosed with JS at younger than 1 year old of age7,8,9,10), and the case of neonatal onset was only one7). The cardinal symptoms of them were unexplainable episodic tachypnea alternating with apnea during neonatal period, abnormal ocular movement, hypotonia, ataxia, and developmental delay. In rare cases, there were renal disease, meningocele, and polydactyly. Most of the patients showed nystagmus, saccadic palsy, or congenital ocular motor apraxia as the abnormal ocular movement, however, one 7-month-old boy had a severe vision loss as Leber's congenital amaurosis8).Ocular and oculomotor involvement is common in JS2,4,11), however, it has variable phenotypes among patients with JS. Colobomas, nystagmus, strabismus, ptosis, and abnormal retinal pigmentation are noted in neonates3). Genetype-phenytype correlations have stressed the predictive value for specific mutations of retinal involvement. Retinal involvement is present in about 80% of patients with AHI1 and CEP290 mutations12). Ocular misalignment is common in JS4,11), because the lesions of cerebellum lead to dysmetric and slow saccades. This patient showed unique eye abnormalities with transient nystagmus, anisocoria and enophthalmos, and eyelid ptosis. Eyelid ptosis should discover the abnormal pupil. When both anisocoria and ptosis are coexistent, one must consider 2 main possible causes: a third nerve palsy (parasympathetic pathway) or a HS (sympathetic pathway)13). In most cases, a third nerve palsy can usually have disconjugate gaze with limitations of eye movements. We had to investigate the possible underlying causes of HS.HS is a problem of the disturbance of the oculosympathetic pathway13), which is bilaterally originating in the hypothalamus, descending ipsilaterally in the reticular formation of the brainstem, eventually synapsing with the second-order neuron in the lower cervical or upper thoracic spinal cord. The second-order neuron enters the sympathetic chain, which then ascends within the soft tissues of the neck, outside of the spinal cord, where it is vulnerable to disruption at various locations. It finally synapses within the superior cervical ganglion to form the third-order neuron. This third-order neuron forms a plexus surrounding the carotid artery and into the cavernous sinus, finally innervating the dilator muscle of the iris and the Müller muscle in the upper and lower eyelids. HS is characterized by a combination of ptosis due to superior tarsal muscle palsy, enophthalmos due to Müller muscle palsy, rising of inferior eyelid due to inferior tarsal muscle palsy, miosis, and anhidrosis of the involved face when there is a defect of the sympathetic nerve route from hypothalamus to the orbit14). The etiology of HS in infants and children differs from the adult population14). Typical causes include birth trauma, neuroblastoma, vascular anomalies of the large arteries, and chest surgery. If there is no history of birth trauma, an acquired HS in children should prompt evaluation for a tumor, particularly paraspinal neuroblastoma. Imaging studies, such as MRI of the head, neck, chest, and abdomen, as well as the measurement of urine catecholamines should be evaluated. Our patient had no underlying cause of HS, compatible with idiopathic HS. These variable ocular phenotypes might be supported by a third nerve palsy and multiplex neuropathological findings of the cerebellar vermis and of several pontine and medullary structures. It may arise from the genetic heterogeneity of JS and related disorders15).In conclusion, we describe an infant of JS with unique eye abnormalities, likely as symptoms of HS. It may suggest that a neonate or young infant with diverse eye abnormalities should be considered for early neuroimaging to evaluate the underlying etiology, although there may be no focal neurologic deficit or significant developmental delay. Furthermore, once a diagnosis of JS has been made, a further evaluation to assess the possibility of multiorgan involvement should be considered in children.
- Acknowledgments
This study was supported by a 2015 research grant from Pusan National University Yangsan Hospital.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology 1968;18:302–303.2. Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 1997;12:423–430.
[Article] [PubMed]3. Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol 1999;14:583–590.
[Article] [PubMed]4. Merritt L. Recognition of the clinical signs and symptoms of Joubert syndrome. Adv Neonatal Care 2003;3:178–186.
[Article] [PubMed]5. Incecik F, Herguner MO, Altunbasak S, Gleeson JG. Joubert syndrome: report of 11 cases. Turk J Pediatr 2012;54:605–611.
[PubMed] [PMC]6. Yachnis AT, Rorke LB. Neuropathology of Joubert syndrome. J Child Neurol 1999;14:655–659.
[Article] [PubMed]7. Lee G, Kim EY, Sun KG, Na KH, Park SY, Kim KS, et al. A case of neonatal onset Joubert syndrome. J Korean Soc Neonatol 2004;11:230–235.8. Yang HK, Yu YS, Hwang JM. Joubert syndrome associated with leber's congenital amaurosis. J Korean Ophthalmol Soc 2008;49:1360–1363.
[Article]9. Jeong HB, Hwang SH, Kim KJ, Hwang YS, Kim SC, Kim IO. Clinical study of symptoms and various anomalies of patients with Joubert syndrome. J Korean Pediatr Soc 1997;40:385–392.10. Jeong JY, Park SJ, Lee YJ, Kim HM, Park SK. A case of joubert syndrome associated with endstage renal disease and meningocele. J Korean Pediatr Soc 1999;42:1311–1316.11. Hodgkins PR, Harris CM, Shawkat FS, Thompson DA, Chong K, Timms C, et al. Joubert syndrome: long-term follow-up. Dev Med Child Neurol 2004;46:694–699.
[Article] [PubMed]12. Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet 2008;51:1–23.
[Article] [PubMed]13. Cahill JA, Ross J. Eye on children: acute work-up for pediatric Horner's syndrome: case presentation and review of the literature. J Emerg Med 2015;48:58–62.
[Article] [PubMed]14. Jeffery AR, Ellis FJ, Repka MX, Buncic JR. Pediatric Horner syndrome. J AAPOS 1998;2:159–167.
[Article] [PubMed]15. Karp N, Grosse-Wortmann L, Bowdin S. Severe aortic stenosis, bicuspid aortic valve and atrial septal defect in a child with Joubert Syndrome and Related Disorders (JSRD): a case report and review of congenital heart defects reported in the human ciliopathies. Eur J Med Genet 2012;55:605–610.
[Article] [PubMed]
Fig. 1
(A) A facial photograph of the baby at the age of 2 months shows left-dominant bilateral ptosis, unilateral enophthalmos of the left eye, high rounded eyebrows, a broad nasal bridge, and a triangular-shaped open mouth. (B) Photograph showing the postoperative status of the ptosis and persisting left-sided enophthalmos at 1 year of age.

Fig. 2
Brain magnetic resonance imaging. (A) Axial T2-fluid-attenuated inversion recovery (FLAIR) image confirming the “molar tooth” appearance of the midbrain with thickened superior cerebellar peduncles (arrows), (B) Coronal T2-weighted image showing agenesis of the cerebellar vermis, (C) Parasagittal T1-weighted image showing a thickened and maloriented superior cerebellar peduncle (open arrowhead), and (D) Bat wing sign in an axial T2-FLAIR image (arrows).
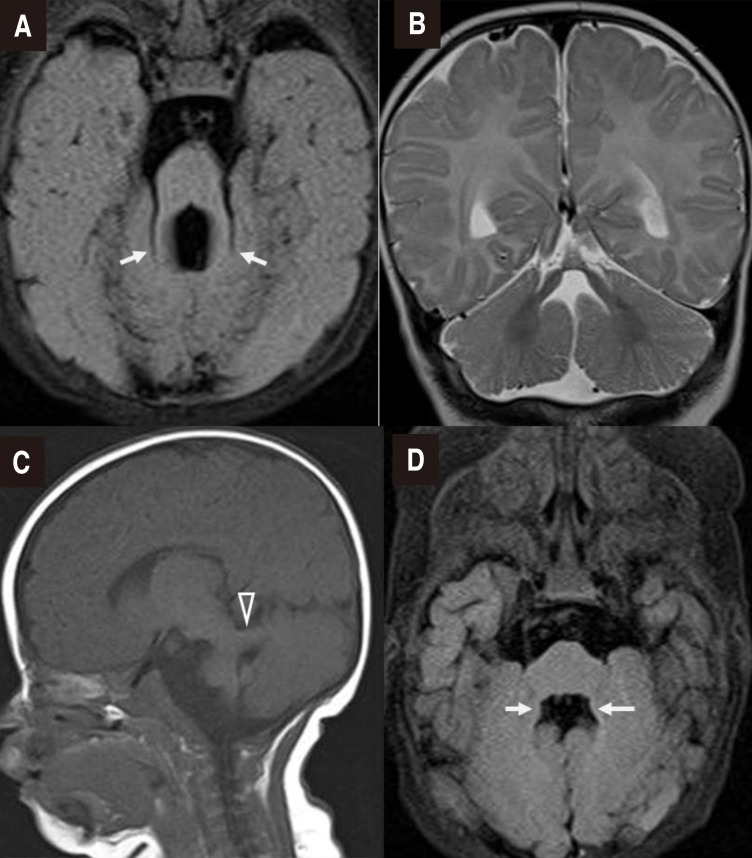
Table 1
Characteristic features associated with Joubert syndrome in the newborn period3)