About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 59(Suppl 1); 2016
A novel BTK gene mutation, c.82delC (p.Arg28 Alafs*5), in a Korean family with X-linked agammaglobulinemia
- Corresponding author: Hyeon-Jong Yang, MD, PhD. Department of Pediatrics, Pediatric Allergy and Respiratory Center, Soonchunhyang University College of Medicine, Seoul, Korea. Tel: +82-2-709-9339, Fax: +82-2-794-5471, pedyang@schmc.ac.kr
- Received September 03, 2014 Revised October 23, 2014 Accepted October 28, 2014
- Abstract
-
X-linked agammaglobulinemia (XLA) is a hereditary humoral immunodeficiency that results from Bruton’s tyrosine kinase (BTK) gene mutations. These mutations cause defects in B-cell development, resulting in the virtual absence of these lymphocytes from the peripheral circulation. Consequently, this absence leads to a profound deficiency of lg all isotypes, and an increased susceptibility to encapsulated bacterial infections. A 15-month-old Korean boy presented with recurrent sinusitis and otitis media after 6 months of age, and had a family history of 2 maternal uncles with XLA. Laboratory tests revealed a profound deficiency of Ig isotypes, and a decreased count of CD19+ B cells in the peripheral circulation. Based on his family history and our laboratory test results, he was diagnosed with XLA. We performed BTK gene analysis of peripheral blood samples obtained from family members to confirm the diagnosis. Mutational analysis revealed a novel hemizygous frameshift mutation (c.82delC, p.Arg28Alafs*5), in the BTK gene. His mother and maternal grandmother were heterozygous carriers of this mutation and his two maternal uncles were hemizygous at the same position. After XLA diagnosis, intravenous immunoglobulin (400 mg/kg, monthly) treatment was initiated; recurrent sinusitis and otitis media were subsequently brought under control. To our knowledge, this is the first reported case of a Korean pedigree with a novel mutation in the BTK gene.
- Introduction
- Introduction
Primary immunodeficiency (PID) is genetically inherited disorder defined as innate or adaptive immunodeficiency, with a clinical result of an increased susceptibility to infection. To date over 150 PID and over 120 related genes have been identified1). XLA is a rare genetic disorder with an estimated prevalence of 1 in 200.000 live births2). The previous nation-wide survey Korean population under 19 years old (2001–2005), has reported that XLA is a 5th common PID with an estimated prevalence of 1.06 person/million3).X-linked agammaglobulinemia (XLA), first described in 1952 by Bruton4), is a hereditary immunodeficiency disease caused by the Bruton tyrosine kinase (BTK) mutations with X-linked recessive inheritance. XLA are characterized by profound deficiency of Ig isotypes resulting from arrest in early B-cell differentiation with marked reduction or absence of mature B cells in the peripheral blood. It increased susceptibility to recurrent encapsulated-bacterial and enteroviral infections leading to chronic respiratory complications, consequently. Thus, BTK gene sequencing is critical tool to confirm the diagnosis of XLA in case with overlapping clinical features like common variable immunodeficiency, transient hypogammaglobulinemia, and other mixed form of humoral immunodeficiency. It will permit to early start optimal treatment before overwhelming infections and end-organ damage occurs5). Up to date, over 1,000 of BTK mutations were known to be associated with XLA, however the present case was identified as a novel BTK mutations which is not yet reported. Thus we report a Korean pedigree with a novel BTK mutation.
- Case report
- Case report
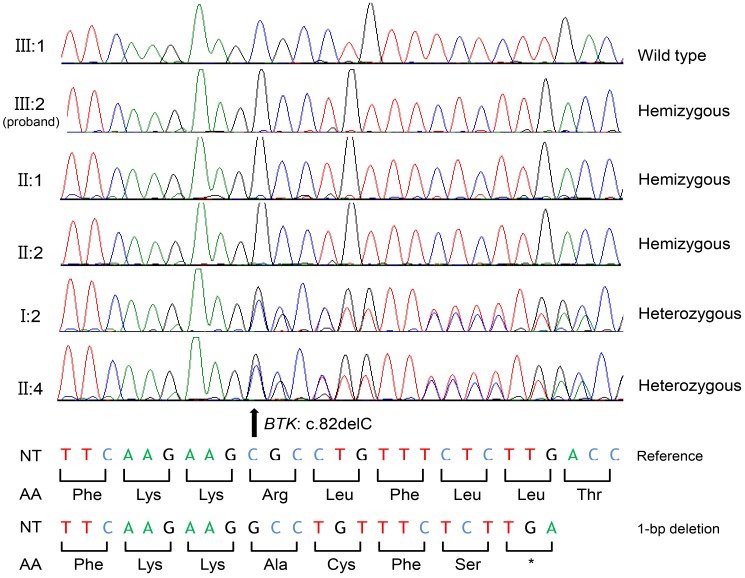
A 15-month-old Korean boy was referred to evaluate the cause of recurrent sinusitis and otitis media. He was born 2,700 g at 40 weeks of gestational age with normal-vaginal delivery, was well up to 6 months of age. After then, he had suffered from recurrent sinusitis and otitis media which failure to treat even though high-dose, oral amoxicillin therapy, consequently. The parent and older brother were healthy, but 2 maternal uncles were previously diagnosed as XLA at the adulthood (Fig. 1).Physical examination showed normal growth and development, body weight was 10.6 kg (25th–50th percentile) and height was 77.2 cm (10th–25th percentile). There were no abnormal morphologic findings except absence of tonsil, purulent postnasal drip and palpable lymph-node. The fluid collection and cloudiness of the ear drum were observed.Laboratory tests revealed as follows: white blood cells, 9,700/µL (neutrophils, 25.0%; lymphocyte, 63.8%); serum IgG, IgA, IgM and IgE levels were 151, 0, 11 mg/dL, and 54.7 KU/L; significantly decreased levels of CD19+ B cells in the peripheral blood (1.1%, 59/µL); the CD4/CD8 T-cell ratio was 2.7:1, respectively.Radiologic examination showed normal finding in chest X-ray, however there was mucosal thickening in both maxillary sinuses and no adenoid shadow in paranasal sinuses views. Based on the family history of XLA, agammaglobulinemia and absent circulating-CD19+ B cells, and absence of adenoid and tonsil with recurrent sinusitis and otitis media, he was diagnosed as XLA. Mutational analysis for BTK gene of the proband and family members was performed to identify the underlying genetic defect. Informed consent was obtained before the start of the study. DNA was extracted from peripheral blood leukocytes. All coding exons with flanking intronic regions of the BTK gene were amplified using the polymerase chain reaction. Sequence chromatograms were compared with the reference sequence of BTK, NM_000061.2. As a result, the proband (III:2) was found to be hemizygous for a 1-bp deletion (c.82delC), which was predicted to result in frameshift at the 28th codon (Arg) and premature termination at the 5th downstream amino acid of the BTK protein (p.Arg28Alafs*5) (Fig. 2). A family study revealed that two maternal uncles (II:1 and II:2) were hemizygous patients and maternal grandmother (I:2), mother (II:4), and cousin (III:4) were heterozygous carriers for the same mutation.After diagnosis of XLA, a scheduled Intravenous Immunoglobulin (IVIG) therapy (400 mg/kg, monthly) was started. The serum level of IgG was reached to target level (or therapeutic level) after 3rd dose of IVIG. The recurrent sinusitis and otitis media were not recurred during 8 months after diagnosis and treatment
- Discussion
- Discussion
The BTK gene is mapped to the midportion of the long arm of X-chromosome at Xq 21.3-Xq 22, and comprises 19 exons which span over 37.5 kb of genomic DNA. In 1993, 2 research groups have identified BTK mutation associating to XLA as molecular basis, and indicated the BTK deficiency interferes with B lineage-specific signal production pathways, which is critical for both early B lineage growth and clonal expansion and mature B lineage survival and activation6,7). The BTK gene encodes a cytoplasmic nonreceptor protein tyrosine kinase, which is a member of the Tec kinase family8). To date, more than 1256 BTK mutations have been identified to be associated with XLA, and are literally spread over the entire domains2). Mutations of the BTK gene are found in approximately 80% of patients with agammaglobulinemia. Mutation types consist of missense mutations (40%), small insertion and deletions (27%), Splice-site mutations (16%) and nonsense mutations (17%)9). To the best of our knowledge, no frameshift mutation affecting Arg28 has been described, while a previously report described several missense mutations at the same nucleotide position. These mutations are listed in BTKbase, a XLA mutation registry, which was established in 1994 (http://structure.bmc.lu.se/idbase/BTKbase/; version 8.53 last updated 30 July, 2014)2). Thus, the mutation described herein is the first frameshift mutation affecting the Arg28 residue of BTK.The BTK protein, a cytoplasmic tyrosine kinase, is involved in signal transduction for B-cell proliferation and development in peripheral circulation6,7). This, is 659 amino-acid residues long, contains 5 signaling terminal domains: pleckstrin homology (PH), Tec homology, Src homology 3, Src homology 2, and catalytic kinase domain, and has diverse partner molecules8). Among these domains, PH is essential for BTK membrane localization, its activation is one of the first steps in antigen-presenting signaling10). The mutation in our patient occurred in PH domain (Arg28) leading to frameshift and premature termination. It is expected to be associated with severe clinical manifestations. Although, no overt genotype-phenotype correlation has been recognized despite much identification of mutation9), even intrafamilial variations and intragenotypical discrepancy have been commonly observed11). The maternal uncles presented a history of recurrent sepsis, even management in the intensive-care units from the early childhood. Thus, our case also expected to be related with severe manifestation if not diagnosed and treated in early. It emphasize that early diagnosis and treatment is the best way to achieve a good clinical outcome, and also suspicion for XLA and gene-study for the differential diagnosis with the clinically overlapping disorders will be essential.Since 1952, intramuscular IgG was first used by Bruton to treat XLA4). There is no doubt that Ig replacement reduces infections, end-organ damage and mortality, use of antibiotics and infection-associated hospitalization as a standard therapy1). The practice guideline provides that IgG replacement initiate 300 to 500 mg/kg every 3 to 4 weeks and alter the dose to achieve a IgG trough level over 500 mg/dL in patient without bronchiectasis to minimize the risk of infection, and over 800 mg/dL might improve chronic pulmonary outcome. The recommended initial infusion-rate is 0.01 mL/kg/min, and can be doubled every 30 minutes until a rate 0.08 mL/kg/min12). It is known that every 100-mg/kg dose increase in 121 mg/dL of IgG, and 100-mg/dL increment of trough IgG level reduce a 27% of incidence of pneumonia13). However, interindividual variability of pharmacokinetics even in same dosage was repeatedly reported. Thus, recent consensus recommends that IgG replacement therapy should be based on the individual clinical response, not simple trough level of IgG14). Our case has kept over 800 mg/dL trough level of IgG after 4th dose of 400 mg/kg with IVIG and has been successfully controlled from bacterial infections. Before introduction of Ig replacement, 75% of XLA patients had developed chronic lung disease at 20 years of age, and 18% had died resulting from infectious complications. The delayed diagnosis and treatment will lead to chronic respiratory complication and serious end-organ damage. Previous literature has reported that most common clinical features of B-cell immunodeficiency in children are recurrent otitis media with markedly decreased or absent tonsils or adenoid tissues, or needs for intravenous antibiotics to clear infections15). Our case also shows absence of tonsil and adenoid tissue with recurrent otitis media failure to treat with standard oral antibiotics. It is thought that those may be early diagnostic clue to suspect XLA. Early diagnosis and therapy, and genetic counseling should be essential to reduce mortality and morbidity. Thus physician should keep a suspicion for this disorder. Furthermore, constructing Korean PID registry system should be essential to much improve acknowledgement.In summary, we report a XLA pedigree with a novel BTK mutation, c.82delC (p.Arg28Alafs*5), leading to frameshift and premature termination. We also emphasize that no tonsil and adenoid tissue with recurrent otitis media and sinusitis which failure to treat with standard antibiotics therapy may be diagnostic clue to suspect XLA.
- Acknowledgments
This work was supported in part by the Soonchunhyang University Research Fund.
- Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Tuerlinckx D, Florkin B, Ferster A, De Schutter I, Chantrain C, Haerynck F, et al. Pneumococcal antibody levels in children with PID receiving immunoglobulin. Pediatrics 2014;133:e154–e162.
[Article] [PubMed]2. Valiaho J, Smith CI, Vihinen M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat 2006;27:1209–1217.
[Article] [PubMed]3. Rhim JW, Kim KH, Kim DS, Kim BS, Kim JS, Kim CH, et al. Prevalence of primary immunodeficiency in Korea. J Korean Med Sci 2012;27:788–793.
[Article] [PubMed] [PMC]5. Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated data from pediatric patients at Severance Hospital. Yonsei Med J 2008;49:28–36.
[Article] [PubMed] [PMC]6. Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science 1993;261:355–358.
[Article] [PubMed]7. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993;72:279–290.
[Article] [PubMed]8. Ponader S, Burger JA. Bruton's tyrosine kinase: from X-linked agammaglobulinemia toward targeted therapy for B-cell malignancies. J Clin Oncol 2014;32:1830–1839.
[Article] [PubMed] [PMC]9. Liu Y, Zhou G, Zhang B, Liu Y. Bruton’s tyrosine kinase: structure and functions, expression and mutations. Gene Technol 2013;2:10610. Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol 2005;23:549–600.
[Article] [PubMed]11. Bykowsky MJ, Haire RN, Ohta Y, Tang H, Sung SS, Veksler ES, et al. Discordant phenotype in siblings with X-linked agammaglobulinemia. Am J Hum Genet 1996;58:477–483.
[PubMed] [PMC]12. Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfus Med Rev 2013;27:171–178.
[Article] [PubMed]13. Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol 2010;137:21–30.
[Article] [PubMed]
Fig. 1
Pedigree of the proband (III:2) with X-linked agammaglobulinemia. Black squares represent clinically affected family members, symbols marked with a point indicate heterozygous carriers of the gene, and arrows indicate the proband (P) in the family. mut+, mutation present; het, heterozygous; hem, hemizygous.
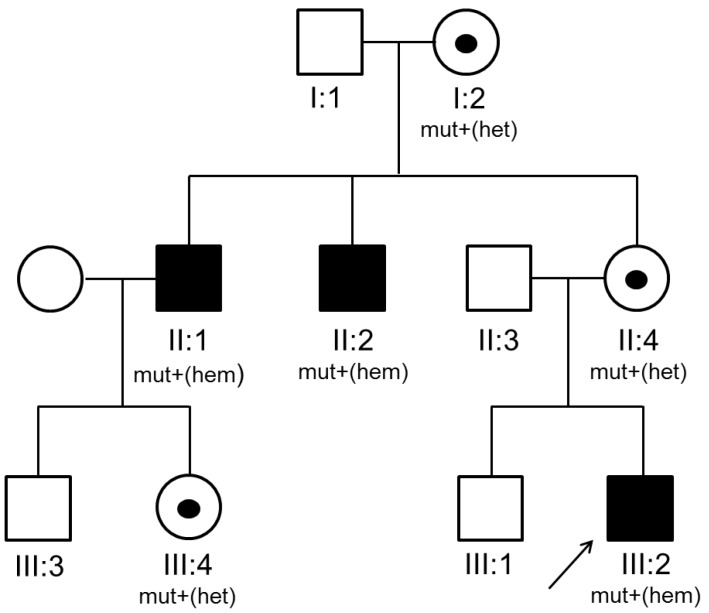
Fig. 2
BTK mutation identified in the proband, and the family members. III:1 is the brother of the proband (III:2), and he possesses the wild type sequence. The proband (III:2) and his maternal uncles (II:1 and II:2) possess a hemizygous allele due to the deletion of the 82nd nucleotide leading to subsequent frameshift mutation at the 28th codon (Arg) predicted to result in premature termination at the 5th downstream amino acid of BTK protein (c.82delC [p.Arg28Alafs*5]). Maternal grandmother (I:2) and mother (II:4) are heterozygous carriers for the same mutation. NT, nucleotide; AA, amino acid; *, stop codon.