 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 63(3); 2020
Genetic classification and confirmation of inherited platelet disorders: current status in Korea
-
Ye Jee Shim, MD, PhD

- Corresponding author: Ye Jee Shim, MD, PhD, Department of Pediatrics, Keimyung University School of Medicine, 1095 Dalgubeol-daero, Dalseo-gu, Daegu 42601, Republic of Korea E-mail: yejeeshim@dsmc.or.kr
- Received January 12, 2019 Revised June 30, 2019 Accepted August 21, 2019
- Abstract
-
Inherited platelet disorders (IPDs), which manifest as primary hemostasis defects, often underlie abnormal bleeding and a family history of thrombocytopenia, bone marrow failure, hematologic malignancies, undefined mucocutaneous bleeding disorder, or congenital bony defects. Wide heterogeneity in IPD types with regard to the presence or absence of thrombocytopenia, platelet dysfunction, bone marrow failure, and dysmegakaryopoiesis is observed in patients. The individual processes involved in platelet production and hemostasis are genetically controlled; to date, mutations of more than 50 genes involved in various platelet biogenesis steps have been implicated in IPDs. Representative IPDs resulting from defects in specific pathways, such as thrombopoietin/MPL signaling; transcriptional regulation; granule formation, trafficking, and secretion; proplatelet formation; cytoskeleton regulation; and transmembrane glycoprotein signaling are reviewed, and the underlying gene mutations are discussed based on the National Center for Biotechnology Information database and Online Mendelian Inheritance in Man accession number. Further, the status and prevalence of genetically confirmed IPDs in Korea are explored based on searches of the PubMed and KoreaMed databases. IPDs are congenital bleeding disorders that can be dangerous due to unexpected bleeding and require genetic counseling for family members and descendants. Therefore, the pediatrician should be suspicious and aware of IPDs and perform the appropriate tests if the patient has unexpected bleeding. However, all IPDs are extremely rare; thus, the domestic incidences of IPDs are unclear and their diagnosis is difficult. Diagnostic confirmation or differential diagnoses of IPDs are challenging, time-consuming, and expensive, and patients are frequently misdiagnosed. Comprehensive molecular characterization and classification of these disorders should enable accurate and precise diagnosis and facilitate improved patient management.
- Introduction
- Introduction
There are 2 important components of hemostasis: primary (platelet response to vessel injury and platelet plug formation) and secondary (generation of fibrin deposition by the coagulation cascade) [1]. Platelets play a key role in the achievement of adequate primary hemostasis through 4 fundamental mechanisms: adhesion, aggregation, secretion, and procoagulant activity [2,3]. Normal platelet adhesion requires the presence of von Willebrand factor (vWF), glycoprotein (GP) receptors on platelets, and the adequate release of platelet granular contents such as adenosine diphosphate (ADP), serotonin, and thromboxane A2 [1]. Thus, familial inherited thrombocytopenia, platelet function disorders, von Willebrand’s disease (vWD), or immune thrombocytopenia show similar primary hemostatic problems [4].However, the diagnostic confirmation and differential diagnosis of IPDs are complicated, time-consuming, and expensive; thus, many patients with IPD are not appropriately diagnosed [4-6]. In most cases, individual megakaryopoiesis, platelet formation, and platelet function are genetically controlled; more than 50 genes associated with IPDs have been identified in recent studies by genome-wide association study and next-generation sequencing [7]. These genes can be classified according to the specific pathway that is disrupted in platelet formation, e.g., the thrombopoietin (THPO)/THPO receptor (MPL) signaling pathway, transcriptional regulation, granule formation/trafficking/secretion, cytoskeleton regulation, and transmembrane protein signaling pathway (Fig. 1) [7].Therefore, in this review, representative IPDs based on genetic classification (Table 1) and the current status of these diseases in Korea (Table 2) will be discussed. In particular, this review focuses on IPDs with thrombocytopenia or thrombocytopathy. The diagnostic sequence and flow in IPD is described in Fig. 2 [4,8].
- THPO/MPL signaling pathway defects
- THPO/MPL signaling pathway defects
- 1. Congenital amegakaryocytic thrombocytopenia
- 1. Congenital amegakaryocytic thrombocytopenia
Congenital amegakaryocytic thrombocytopenia (CAMT) (OMIM #604498) is a rare autosomal recessive IPD with severe thrombocytopenia from infancy due to MPL gene mutation and high levels of serum THPO [9]. The case of a genetically confirmed Korean patient with CAMT harboring a novel mutation in the MPL gene was recently reported [10]. CAMT is characterized by megakaryocytopenia in the bone marrow, thrombocytopenia with normal platelet size, and progressive bone marrow failure to aplastic anemia [9]. Approximately 10%–30% of patients with CAMT have orthopedic/neurological abnormalities or intracranial hemorrhage [6]. The THPO receptor, encoded by the MPL gene, promotes megakaryocyte growth and proliferation and may play a role in maintaining hematopoietic stem cells [9,11]. CAMT with bone marrow failure to aplastic anemia is best managed by a hematopoietic stem cell transplantation (HSCT) [12].
- Defects in transcriptional regulation
- Defects in transcriptional regulation
- 1. X-linked thrombocytopenia with or without dyserythropoietic anemia
- 1. X-linked thrombocytopenia with or without dyserythropoietic anemia
X-linked thrombocytopenia with or without dyserythropoietic anemia (XLTDA) (OMIM #300367) is a rare X-linked recessive IPD with an unknown prevalence; only a few cases have been reported worldwide [13-16]. No Korean cases of XLTDA associated with GATA1 have been reported to date. The GATA1 gene encodes a crucial transcription factor, GATA-binding factor 1, involved in the development of erythrocytes and megakaryocytes in the early stage of hematopoiesis [14]. Depending on the specific GATA1 mutation, macrothrombocytopenia, variable severity of anemia, hemolysis, and hypercellular marrow with erythroid dysplasia can occur [2].- 2. Jacobsen syndrome and Paris-Trousseau syndrome
- 2. Jacobsen syndrome and Paris-Trousseau syndrome
Jacobsen syndrome (OMIM #147791) and Paris-Trousseau syndrome (OMIM #188025) are rare autosomal dominant IPDs associated with the deletion of chromosome 11q [17,18]. A few Korean cases have been reported [19-21]. These diseases share several similar phenotypes (dysmorphic features, intellectual disability) and macrothrombocytopenia with giant α-granules due to the fusion of smaller organelles [17,22]. Chromosome 11q23.3 includes the FLI1 gene, which encodes an essential transcription factor for megakarypoiesis [23]. Hemizygous deletion of the FLI1 gene prevents progenitor cells from undergoing the normal differentiation process, thereby generating a large population of small immature megakaryocytes that undergo lysis [23].- 3. Radioulnar synostosis with amegakaryocytic thrombocytopenia
- 3. Radioulnar synostosis with amegakaryocytic thrombocytopenia
Radioulnar synostosis with amegakaryocytic thrombocytopenia (RUSAT), consisting of RUSAT1 (OMIM #605432) and RUSAT2 (OMIM #616738) types, is a recently identified and introduced autosomal dominant IPD characterized by progressive bone marrow failure and pancytopenia requiring HSCT [24,25]. Only a few families with these conditions have been reported worldwide [2], and no Korean cases have been described in the literature. These syndromes involve amegakaryocytic thrombocytopenia and skeletal defects (limited forearm pronation/supination due to proximal fusion of the radius and ulna) [24,25]. RUSAT1 and RUSAT2 are caused by variants in HOXA11 and MECOM genes, respectively [24,25]. The protein encoded by the HOXA11 gene is a DNA-binding transcription factor that regulates gene expression, morphogenesis, and differentiation [24]. The oncoprotein EVI1, encoded by the MECOM gene, is also a transcriptional regulator involved in hematopoiesis and stem cell self-renewal in humans [26]. In mice, MECOM protein is expressed at high levels in the embryonic limb bud, suggesting that this gene is also involved in limb development [27].- 4. Thrombocytopenia-absent radius syndrome
- 4. Thrombocytopenia-absent radius syndrome
Thrombocytopenia-absent radius (TAR) syndrome (OMIM #274000), also known as chromosome 1q21.1 deletion syndrome, is a rare autosomal recessive IPD with congenital hypomegakaryocytic thrombocytopenia and bilateral radial aplasia [28]. A few Korean cases have been reported to date [29,30]. A mutation in RBM8A gene was recently shown to cause TAR syndrome [31]. RBM8A has several important cellular functions in the production of other proteins by facilitating mRNA transport.31) In TAR syndrome, thrombocytopenia becomes less severe over time; the platelet levels reportedly normalized in some cases [2,6,28]. Some patients also have other congenital anomalies, e.g., lower-limb anomalies, cow’s milk intolerance, renal anomalies, cardiac anomalies, intracranial vascular malformation, facial hemangioma, sensorineural hearing loss, and scoliosis [28]. A skeletal evaluation and bone marrow study are important in thrombocytopenia such as RUSAT or TAR. Two diseases can emphasize that a thorough orthopedic examination is necessary for patients with thrombocytopenia.
- Defects in granule formation, trafficking, or secretion
- Defects in granule formation, trafficking, or secretion
- 1. Hermansky-Pudlak syndrome (dense granule disorder)
- 1. Hermansky-Pudlak syndrome (dense granule disorder)
Hermansky-Pudlak syndrome (HPS) is a rare heterogeneous autosomal recessive IPD group with several genetic defects, including mutations in HPS1 (OMIM #203300), AP3B1 (HPS2, OMIM #608233), HPS3 (OMIM #614072), HPS4 (OMIM #614073), HPS5 (OMIM #614074), HPS6 (OMIM #614075), DTNBP1 (HPS7; OMIM #614076), BLOC1S3 (OMIM #614077), and BLOC1S6 (OMIM #614171) [7,32]. HPS is a disease group of defects in lysosomes, melanosomes, and granule biogenesis [33]. These conditions share features such as oculocutaneous albinism and bleeding symptoms due to platelet dysfunction associated with dense granule defects [2,6,32]. Owing to the presence of dense granule defects, HPS platelets result in aggregation defects with the absence of second-wave aggregation in response to adrenaline [2]. Proteins encoded by the HPS gene are involved in intracellular vesicle transport, protein sorting, and vesicle docking/fusion [2,33]. Additional manifestations, including pulmonary fibrosis, granulomatous colitis, neutropenia, or immunodeficiency, can occur according to mutation type [2,6,32]. No Korean cases of HPS have been reported; however, HPS is a common genetic disorder in Puerto Rico, affecting 1 in 800 individuals, with more than 500 reported cases [34].- 2. Chediak-Higashi syndrome (dense granule disorder)
- 2. Chediak-Higashi syndrome (dense granule disorder)
Chediak-Higashi syndrome (CHS) (OMIM #214500) is a rare autosomal recessive IPD characterized by ocular cutaneous hypopigmentation, platelet dysfunction, immunodeficiency due to abnormal natural killer cell function, and peripheral neuropathy associated with mutations in the lysosomal trafficking regulator (LYST) gene [35,36]. Thus, CHS disrupts the structure and function of lysosomes and related structures in cells. HPS and CHS share common clinical manifestations (oculocutaneous albinism and platelet dysfunction due to a dense granule defect) but can be differentially diagnosed by the presence of large peroxidase-positive cytoplasmic granules in neutrophils and more severe symptoms at an earlier age in the case of CHS [35]. Few Korean CHS cases have been reported to date [37].
- Defects in proplatelet formation and cytoskeleton regulation
- Defects in proplatelet formation and cytoskeleton regulation
- 1. MYH9-related disorders
- 1. MYH9-related disorders
MYH9-related disorders (OMIM #155100) are autosomal dominant IPDs including May-Hegglin anomaly (MHA), Fechtner syndrome (FTNS), Sebastian syndrome (SBS), and Epstein syndrome (ES) [38]. All of these syndromes are associated with the MYH9 gene, which encodes nonmuscle myosin heavy chain IIa (NMMHC-IIA), a component of the contractile cytoskeleton in many tissues, including megakaryocytes, platelets, leukocytes, and the kidneys [38,39]. The altered distribution of NMMHC-IIA within platelets can be measured by immunohistochemistry or immunofluorescence [39]. Thrombocytopenia resulting from defective megakaryocytosis and typical cytoplasmic basophilic inclusion bodies of leukocytes in MYH9-related disorders is caused by the aggregation of abnormal NMMHC-IIA [6]. According to several studies, MHA, FTNS, SBS, and ES are not separate entities but a single disorder with a wide spectrum of clinical symptoms, e.g., from mild macrothrombocytopenia to the more severe form with sensory neural hearing loss, renal failure, and cataracts [40-42]. Platelet counts are typically in the range of 20–130×109/L, with elevated mean platelet volume and mild bleeding symptoms [6,42]. To date, several Korean MYH9-related disorders confirmed by genetic testing have been reported [43-48].- 2. Wiskott-Aldrich syndrome and X-linked thrombocytopenia
- 2. Wiskott-Aldrich syndrome and X-linked thrombocytopenia
Wiskott-Aldrich syndrome (WAS) (OMIM #301000) is a rare X-linked recessive IPD, with an incidence of approximately 1–4 per 1,000,000 live male births [2,6]. Although the prevalence of most IPDs in Korean populations has not yet been determined, the prevalence of Korean WAS is 0.33 per 1,000,000 individuals based on an epidemiological study performed using data of the national registry for primary immunodeficiencies [49]. The WAS protein (WASP), expressed in hematopoietic cell lineages, has a fundamental role in signal transduction from receptors on the cell surface to the actin cytoskeleton [2,6,50,51]. Thus, aberrant WASP expression results in defective proplatelet formation and diverse clinical manifestations of WAS, e.g., microthrombocytopenia, eczema, frequent infection due to immunodeficiency, and even malignancy [2,6,50,51]. Microthrombocytopenia is the characteristics of WAS, and present at birth and varies between 5 and 50×109/L [2]. Malignancies occur in 13% of patients with WAS at a mean 9.5 years of age [52].X-linked thrombocytopenia (XLT) (thrombocytopenia 1; OMIM #313900), the milder variant, is characterized by thrombocytopenia with transient eczema and absent or minimal immunodeficiency [51]. WAS gene-associated XLT can be misdiagnosed as chronic ITP because it is milder without frequent infection or eczema [6]. Several cases of Korean patients with WAS or XLT confirmed by genetic testing have been reported to date [53-60]. The results of hematopoietic stem cell transplantation in Korean patients with WAS have also been reported [54,58,61]. XLT is difficult to be differentially diagnosed clinically from XLTDA (OMIM #300367). In the case of XLT, there may be a family history of WAS, which will help with differentiation between the 2 diseases. In addition, platelet size (large in XLTDA, small in XLT) is helpful for differentiating between the 2 diseases.
- Transmembrane GP signaling pathway defects
- Transmembrane GP signaling pathway defects
- 1. Glanzmann thrombasthenia
- 1. Glanzmann thrombasthenia
Glanzmann thrombasthenia (GT) (OMIM #273800) is a rare autosomal recessive IPD with a prevalence of approximately 1 in every 1,000,000 individuals, although a higher prevalence was observed in some populations owing to increased rates of consanguineous marriages [62]. GT typically develops with loss-of-function variants of the ITGB2A or ITGB3 genes encoding GPIIb and GPIIIa, respectively [63]. However, very rare gain-of-function (GOF) variants in the ITGB3 gene have also been reported, resulting in enhanced fibrinogen binding and severe bleeding [64]. To date, several Korean cases of GT have been diagnosed clinically or confirmed by flow cytometry; of them, some were further confirmed by genetic analysis [65]. In patients with GT, there are quantitative and/or qualitative defects in the platelet GPIIb/IIIa complex at binding sites for fibrinogen, vWF, and fibronectin [66,67]. Thus, patients with GT exhibit normal platelet count/morphology but severely diminished platelet aggregation in response to ADP, epinephrine, serotonin, thrombin, and collagen. Platelet function analyser-100 (PFA-100) measurements are significantly abnormal in cases of GT, with prolonged closure times on ADP/collagen and adrenaline/collagen cartridges [2]. Impaired platelet agglutination in patients with GT is corrected by ristocetin, which induces the binding of vWF with GPIb/IX [2]. The application of flow cytometry using antibodies to GPIIb (CD41)/GPIIIa (CD61) is also useful for the diagnosis of GT [2].- 2. Bernard-Soulier syndrome
- 2. Bernard-Soulier syndrome
Bernard-Soulier syndrome (BSS) (OMIM #231200) is a rare IPD that is typically transmitted in an autosomal recessive manner; its prevalence is estimated to be less than 1 in 1,000,000 individuals [6]. To date, fewer than 1,000 BSS cases have been reported and studied worldwide [2,6,68]. No Korean BSS cases have been published to date. BSS is characterized by low platelet counts, typically ranging from less than 30 to 200×109/L, and the presence of large platelets (macrothrombocytopenia) in a peripheral blood smear [6,69]. These large platelets are associated with the abnormal development of platelet membranes due to GPIba abnormalities [70]. In patients with BSS, platelets have defective surface expression of GPIb-IX-V, which acts as a vWF receptor on platelets, owing to mutations in the GP1BA, GP1BB, or GP9 genes [2,69,71]. As a result, platelets in patients with BSS cannot adhere to the vascular subendothelium and are unable to be agglutinated by ristocetin [68]. In a study by the International Consortium for BSS, 211 BSS families had mutations in the GP1BA (28%), GP1BB (28%), or GP9 (44%) genes [68]. The closure time in PFA-100 is significantly prolonged in BSS on both cartridges [2]. The detection of defects in the GPIb-IX-V complex by antibodies to GPIb (CD42b) on flow cytometry is also useful for making the diagnosis of BSS [72].- 3. Pseudo-vWD
- 3. Pseudo-vWD
Pseudo-vWD (OMIM #177820), also known as platelet-type vWD or bleeding disorder platelet-type 3, results from GOF variants in the GP1BA gene [73]. Pseudo-vWD is an autosomal dominant genetic disorder of the platelets, and genetic tests of the vWF gene are normal [73]. GOF variants in GP1BA affect the qualitatively altered GPIb receptor, which then shows increased affinity to vWF [74]. Accordingly, large platelet aggregates and high-molecular-weight vWF multimers are removed from circulation, resulting in thrombocytopenia and altered vWF multimers. Ristocetin cofactor activity and altered vWF multimers are similar to characteristics of vWD type 2B, a disorder of GOF variants in the vWF gene resulting in altered function of the A1 domain of vWF [75]. In both cases, increased affinity of the GP1b receptor and vWF A1 domain results in thrombocytopenia and abnormal vWF multimers. Thus, DNA analysis is required for differential diagnosis [7].- 4. 22q11 deletion syndromes
- 4. 22q11 deletion syndromes
The 22q11.21 deletion syndromes, including DiGeorge syndrome (OMIM #188400) and velocardiofacial syndrome (OMIM #192430), may be related to BSS phenotype if the remaining single GPIBB gene contains an independently inherited mutation [76]. These 22q11 deletion syndromes are autosomal dominantly inherited disorders with an estimated incidence of 1 in 4,000 births and are characterized by congenital heart disease, specific facial features, thymic aplasia, frequent infections, cleft palate, hypocalcemia, hypoparathyroidism, learning disabilities, and developmental delay [77]. If patients must undergo heart surgery because of congenital heart disease associated with 22q11.2 deletion syndrome, physicians should be aware of the serious bleeding risk in these patients [78].
- Conclusions
- Conclusions
In the last few decades, researchers have elucidated many of the etiologies of IPDs. The genes involved in a variety of IPDs have been identified, and molecular characterization of these disorders is underway. This information enables a more accurate understanding of IPDs. To enable more accurate and definitive diagnostics, advanced genetic approaches, including next-generation sequencing, are required for patients with suspected IPD [79,80]. Although the NGS panel for IPDs has not yet been commercialized and popularized in Korea, interest in IPDs is increasing. As described in Table 2, Korean Pediatric Hematology Oncology Group recently attempted the genetic confirmation of IPDs using targeted exome sequencing as a multicenter study. Although this study has only just started, its findings are expected to be useful for future large-scale studies and the establishment of a Korean IPD registry. It also may be useful for the future development of an NGS panel for IPDs in Korea.
- Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Fig. 1.
Identified genes associated with inherited platelet disorders. Each of the genes can be classified according to the specific pathway that is disrupted in megakaryopoiesis or platelet formation. HSC, hematopoietic stem cell; MK, megakaryocyte; GP, glycoprotein; GPCR, G-protein-coupled receptor. Adapted from Lentaigne et al., Blood 2016;127: 2814-23 [7].
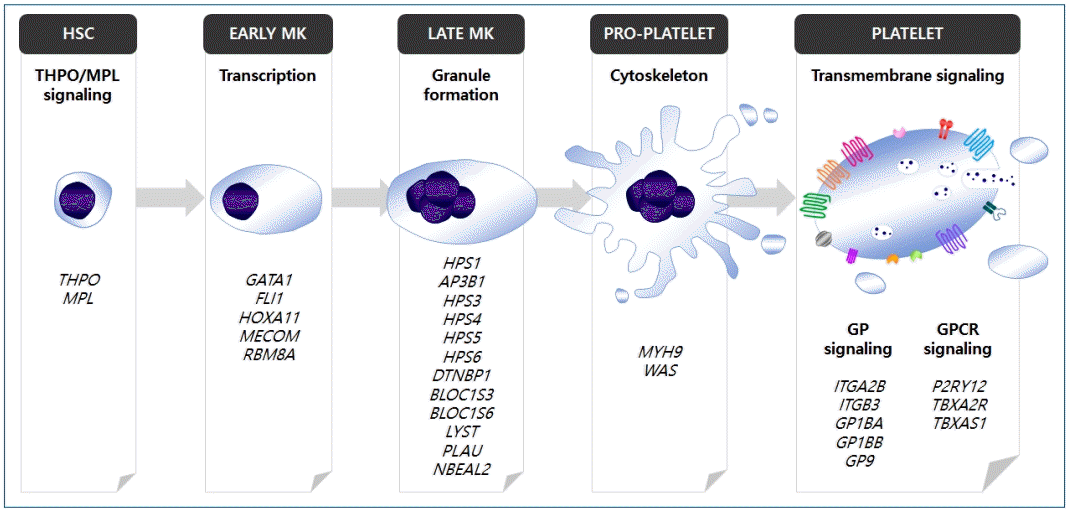
Fig. 2.
Diagnostic sequence and flow in inherited platelet disorders (IPDs). BM, bone marrow; WAS, Wiskott-Aldrich syndrome; XLT, X-linked thrombocytopenia; CAMT, congenital amegakaryocytic thrombocytopenia; RUSAT, radioulnar synostosis with amegakaryocytic thrombocytopenia; TAR, thrombocytopenia-absent radius; QPD, Quebec platelet disorder; XLTDA, X-linked thrombocytopenia with or without dyserythropoietic anemia; MYH9, MYH9-related disorders; BSS, Bernard-Soulier syndrome; vWD, von Willebrand disease; 22q11 del, 22q11 deletion syndrome; GPS, gray platelet syndrome; JS, Jacobsen syndrome; PTS, Paris-Trousseau syndrome; GT, Glanzmann thrombasthenia; HPS, Hermansky-Pudlak syndrome; CHS, Chediak-Higashi syndrome.

Table 1.
Genetic classification of inherited platelet disorders by megakaryopoiesis or platelet formation
| Process of plate formationlet | Gene | Locus | Protein | Phenotype (OMIM number) | Inheritance | Clinical characteristics |
|---|---|---|---|---|---|---|
| THPO/MPL signaling pathway | THPO | 3q27.1 | Thrombopoietin | Thrombocythemia 1 (OMIM #187950) [81] | AD | Familial thrombocytosis |
| Increased megakaryocytes in bone marrow | ||||||
| MPL | 1p34.2 | Thrombopoietin receptor | Congenital amegakaryocytic thrombocytopenia (OMIM #604498) [9] | AR | Absence of megakaryocyte in bone marrow | |
| Thrombocytopenia | ||||||
| Normal platelet size | ||||||
| Elevated serum thrombopoietin | ||||||
| Progressive bone marrow failure to aplastic anemia | ||||||
| Thrombocythemia 2 (OMIM #601977) [82] | AD | Familial thrombocytosis | ||||
| Increased megakaryocytes in bone marrow | ||||||
| Transcriptional regulation | GATA1 | Xp11.23 | GATA-binding factor 1 | X-linked thrombocytopenia with or without dyserythropoietic anemia (OMIM #30036) [13-15] | XR | Thrombocytopenia |
| Large platelet size | ||||||
| Variable severity of anemia and hemolysis | ||||||
| Dyserythropoietic anemia | ||||||
| FLI1 | 11q24.3 | Friend leukemia integration 1 transcription factor | Jacobsen syndrome (OMIM #147791) [22,23] | AD | Thrombocytopenia | |
| Paris-Trousseau type thrombocytopenia (OMIM #188025) [17,22] | Large platelet size | |||||
| Chromosome 11q deletion syndrome [18] | Giant α-granules due to fusion of small organelles | |||||
| Dysmegakaryocytopoiesis in bone marrow | ||||||
| HOXA11 | 7p15.2 | Homeobox protein Hox-A11 | Radioulnar synostosis with amegakaryocytic thrombocytopenia 1 (OMIM #605432) [24] | AD | Amegakaryocytic thrombocytopenia | |
| Radioulnar synostosis | ||||||
| MECOM | 3q26.2 | MDS1 and EVI1 complex locus protein | Radioulnar synostosis with amegakaryocytic thrombocytopenia 2 (OMIM #616738) [25] | AD | Amegakaryocytic thrombocytopenia | |
| Radioulnar synostosis | ||||||
| RBM8A | 1q21.1 | RNA binding motif protein 8A | Thrombocytopenia-absent radius syndrome (OMIM #274000) [31] | AR | Amegakaryocytic thrombocytopenia | |
| Absence of radius | ||||||
| Granule formation, trafficking, or secretion | HPS1 | 10q24.2 | Hermansky-Pudlak syndrome [33,34] (OMIM #203300, #608233, #614072, #614073, #614074, #614075, #614076, #614077, #614171) | AR | Normal platelet count | |
| AP3B1 | 5q14.1 | Platelet aggregation defect or abnormal response | ||||
| HPS3 | 3q24 | |||||
| HPS4 | 22q12.1 | Platelet dense granule defect | ||||
| HPS5 | 11p15.1 | Oculocutaneous albinism | ||||
| HPS6 | 10q24.32 | Congenital neutropenia | ||||
| DTNBP1 | 6p22.3 | Pulmonary fibrosis | ||||
| BLOC1S3 | 19q13.32 | Granulomatous colitis | ||||
| BLOC1S6 | 15q21.1 | |||||
| LYST | 1q42.3 | Lysosomal trafficking regulator | Chediak-Higashi syndrome (OMIM #214500) [35] | AR | Normal platelet count | |
| Platelet aggregation defect or abnormal | ||||||
| response | ||||||
| Platelet dense granule defect | ||||||
| Oculocutaneous albinism | ||||||
| Severe immunological deficiency | ||||||
| Lack of NK cell function | ||||||
| PLAU | 10q22.2 | Urinary plasminogen activator | Quebec platelet disorder (OMIM #601709) [83] | AD | AD Gain-of-function defect in fibrinolysis | |
| Thrombocytopenia or normal platelet count | ||||||
| Degradation of platelet α-granule contents | ||||||
| Platelet aggregation defect or abnormal response | ||||||
| NBEAL2 | 3p21.31 | Beige and Chediak-Higashidomain protein | Gray platelet syndrome (OMIM #139090) [84] | AR | Thrombocytopenia | |
| Platelet α-granule defect | ||||||
| Large-sized platelets | ||||||
| Platelet aggregation defect | ||||||
| Gray platelets on light microscopy of Wright-stain | ||||||
| Myelofibrosis | ||||||
| Cytoskeleton regulation | MYH9 | 22q12.3 | Nonmuscle myosin heavy chain IIa | MYH9-related thrombocytopenia syndromes (OMIM #155100) [37,40-42] | AD | Thrombocytopenia or normal platelet count |
| Large-sized platelets | ||||||
| Nephritis | ||||||
| Sensorineural hearing loss | ||||||
| WAS | Xp11.23 | WAS protein | Wiskott-Aldrich syndrome (OMIM #301000) [50] | XR | Thrombocytopenia | |
| Small-sized platelets | ||||||
| Immunodeficiency | ||||||
| Eczema | ||||||
| Thrombocytopenia 1 (OMIM #313900)= X-linked thrombocytopenia [51] | XR | Thrombocytopenia | ||||
| Small-sized platelets | ||||||
| Without profound immunodeficiency | ||||||
| Defects of white blood cell cytoskeleton | ||||||
| Transient eczema | ||||||
| Transmembrane GP signaling pathway | ITGA2B | 17q21.31 | GP Iib | Glanzmann thrombasthenia (OMIM #273800) [63] | AR | Normal platelet count |
| ITGB3 | 17q21.32 | GP IIIa | Platelet aggregation defect | |||
| GP1BA | 17p13.2 | GP Ib | Bernard-Soulier syndrome (OMIM #231200) [68,69] | AR | Thrombocytopenia or normal platelet count | |
| GP1BB | 22q11.2 | GP Ib | Large-sized platelet | |||
| GP9 | 13q21.3 | GP IX | Platelet adhesion defect | |||
| GP1BA | 17p13.2 | GP Ib | Pseudo-von Willebrand disease (OMIM #177820) [73,74] | AD | Thrombocytopenia | |
| Large-sized platelet | ||||||
| Platelet adhesion defect | ||||||
| GP1BB | 22q11.21 | GP Ib | 22q11 deletion syndrome [76-78] | AD | Thrombocytopenia or normal platelet count | |
| - De George syndrome (OMIM #188400) | Large-sized platelet | |||||
| - Velocardiofacial syndrome (OMIM #192430) | Platelet adhesion defect | |||||
| GPCR signaling pathway | P2RY12 | 3q25.1 | ADP receptor | Bleeding disorder, platelettype, 8 (OMIM #609821) [85] | AR | Platelet aggregation defect |
| TBXA2R | 19p13.3 | Thromboxane A2 receptor | Bleeding disorder, platelettype, 13 (OMIM #614009) [86] | AD | Platelet aggregation defect | |
| TBXAS1 | 7q34 | Thromboxane synthase | Thromboxane synthase deficiency (OMIM #614158) [87] | AD | Platelet aggregation defect | |
| Ghosal hematodiaphyseal syndrome (OMIM #231095) [88] | AR | Platelet aggregation defect | ||||
| Other | ANO6 | 12q12 | Transmembrane protein 16F | Scott syndrome (OMIM #262890) [89] | AR | Impaired surface exposure of phosphatidylserine of platelets |
Table 2.
Current status of genetic confirmation of inherited platelet disorders in Korea
| Phenotype | Genetically confirmed Korean IPD patients | Diagnostic method |
|---|---|---|
| Congenital amegakaryocytic thrombocytopenia Jacobsen syndrome | 1 Male infant patient reported by Chung et al. [10] | Direct sequencing of MPL |
| 1 Male premature neonate reported by Noh et al. [19] | Karyotype | |
| 1 Female prenatal case reported by Yoon et al. [21] | Karyotype through amniocentesis | |
| 1 Female infant reported by Shin et al. [20] | Chromosomal microarray and Karyotype | |
| Thrombocytopenia-absent radius syndrome | 1 Male infant reported by Kim et al. [29] | Chromosomal microarray |
| 1 Prenatal case reported by We et al. [30] | Fetal blood analysis | |
| MYH9-related thrombocytopenia syndromes | 1 Male adult patient reported by Jang et al. [44] | Direct sequencing of MYH9 |
| 5 Families (20 patients) reported by Kook et al. [45] | Direct sequencing of MYH9 | |
| 1 Family reported from Lee, et al. [46] | Direct sequencing of MYH9 | |
| 1 Female adult patient reported by Oh et al. [47] | Direct sequencing of MYH9 | |
| 7 Patients (5 male and 2 female) reported by Han et al. [43] | Direct sequencing of MYH9 | |
| 1 Female pediatric patient reported by Park et al. [48] | Direct sequencing of MYH9 | |
| 1 Male patient by K-PHOG study (unpublished data)a) | Targeted exome sequencing | |
| Wiskott-Aldrich syndrome | 1 Male infant reported by Hwang et al. [90] | PCR-SSCP and direct sequencing of WAS |
| 1 Male pediatric patient reported by Baek et al. [91] | Direct sequencing of WAS | |
| 2 Male infants reported by Jo et al. [53] | Direct sequencing of WAS | |
| 1 Male pediatric patient reported by Kang et al. [54] | PCR-SSCP of WAS | |
| 1 Male infant reported by Kim et al. [55] | Direct sequencing of WAS | |
| 2 Families reported by Kim, et al. [56] | Direct sequencing of WAS | |
| 1 Male infant reported by Lee et al. [58] | Direct sequencing of WAS | |
| 1 Family reported by Park et al. [59] | PCR-SSCP and direct sequencing of WAS | |
| 1 Male adolescent patient reported by Yoon et al. [60] | Direct sequencing of WAS | |
| X-linked thrombocytopenia | 1 Male pediatric patient reported by Lee et al. [57] | Direct sequencing of WAS |
| 1 Male pediatric patient reported by Yoon et al. [60] | Direct sequencing of WAS | |
| Glanzmann thrombasthenia | 4 Patients reported by Park et al. [65] | Direct sequencing of ITGB3 and ITGA2B |
| 7 Patients by K-PHOG study (unpublished data)a) | Targeted exome sequencing |
- References
- 1. Gale AJ. Continuing education course #2: current understanding of hemostasis. Toxicol Pathol 2011;39:273–80.
[Article] [PubMed]2. Bolton-Maggs PH, Chalmers EA, Collins PW, Harrison P, Kitchen S, Liesner RJ, et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol 2006;135:603–33.
[Article] [PubMed]3. Nurden AT, Freson K, Seligsohn U. Inherited platelet disorders. Haemophilia 2012;18 Suppl 4:154–60.
[Article] [PubMed]4. Lambert MP. What to do when you suspect an inherited platelet disorder. Hematology Am Soc Hematol Educ Program 2011;2011:377–83.
[Article] [PubMed]5. Gresele P, Harrison P, Bury L, Falcinelli E, Gachet C, Hayward CP, et al. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J Thromb Haemost 2014;12:1562–9.
[Article] [PubMed]6. D'Andrea G, Chetta M, Margaglione M. Inherited platelet disorders: thrombocytopenias and thrombocytopathies. Blood Transfus 2009;7:278–92.
[PubMed] [PMC]7. Lentaigne C, Freson K, Laffan MA, Turro E, Ouwehand WH, BRIDGE-BPD Consortium and the ThromboGenomics Consortium. Inherited platelet disorders: toward DNA-based diagnosis. Blood 2016;127:281423
[Article]8. Israels SJ, El-Ekiaby M, Quiroga T, Mezzano D. Inherited disorders of platelet function and challenges to diagnosis of mucocutaneous bleeding. Haemophilia 2010;16 Suppl 5:152–9.
[Article] [PubMed]9. Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A, et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 2001;97:139–46.
[Article] [PubMed]10. Chung HS, Koh KN, Kim HJ, Kim HJ, Lee KO, Park CJ, et al. A novel nonsense mutation in the MPL gene in congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer 2011;56:304–6.
[Article] [PubMed]11. Al-Qahtani FS. Congenital amegakaryocytic thrombocytopenia: a brief review of the literature. Clin Med Insights Pathol 2010;3:25–30.
[Article] [PubMed] [PMC]12. Al-Ahmari A, Ayas M, Al-Jefri A, Al-Mahr M, Rifai S, El-Solh H. Allogeneic stem cell transplantation for patients with congenital amegakaryocytic thrombocytopenia (CAT). Bone Marrow Transplant 2004;33:829–31.
[Article] [PubMed]13. Freson K, Devriendt K, Matthijs G, Van Hoof A, De Vos R, Thys C, et al. Platelet characteristics in patients with X-linked macrothrombocytopenia because of a novel GATA1 mutation. Blood 2001;98:85–92.
[Article] [PubMed]14. Mehaffey MG, Newton AL, Gandhi MJ, Crossley M, Drachman JG. X-linked thrombocytopenia caused by a novel mutation of GATA-1. Blood 2001;98:2681–8.
[Article] [PubMed]15. Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet 2000;24:266–70.
[Article] [PubMed] [PMC]16. Yu C, Niakan KK, Matsushita M, Stamatoyannopoulos G, Orkin SH, Raskind WH. X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood 2002;100:2040–5.
[Article] [PubMed] [PMC]17. Favier R, Jondeau K, Boutard P, Grossfeld P, Reinert P, Jones C, et al. Paris-Trousseau syndrome: clinical, hematological, molecular data of ten new cases. Thromb Haemost 2003;90:893–7.
[Article] [PubMed]18. Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, et al. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A 2004;129A:51–61.
[Article] [PubMed]19. Noh JH, Park IS, Lee HK, Kim YC. A case of Jacobsen syndrome. J Korean Soc Neonatol 2002;9:211–4.20. Shin J, Kim G, Lee R, Jung N, Shim YJ, Ha JS. A case of Jacobsen syndrome presenting with a huge cephalhematoma and thrombocytopenia after birth. Clin Pediatr Hematol Oncol 2018;25:50–60.
[Article]21. Yoon JH, Kim SR, Lee WI, Do IG, Lee BY, Lee SK, et al. A case of prenatally diagnosed Jacobsen syndrome. Korean J Obstet Gynecol 2005;48:1358–61.22. Krishnamurti L, Neglia JP, Nagarajan R, Berry SA, Lohr J, Hirsch B, et al. Paris-Trousseau syndrome platelets in a child with Jacobsen's syndrome. Am J Hematol 2001;66:295–9.
[Article] [PubMed]23. Raslova H, Komura E, Le Couédic JP, Larbret F, Debili N, Feunteun J, et al. FLI1 monoallelic expression combined with its hemizygous loss underlies Paris-Trousseau/Jacobsen thrombopenia. J Clin Invest 2004;114:77–84.
[Article] [PubMed] [PMC]24. Thompson AA, Nguyen LT. Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet 2000;26:397–8.
[Article] [PubMed]25. Niihori T, Ouchi-Uchiyama M, Sasahara Y, Kaneko T, Hashii Y, Irie M, et al. Mutations in MECOM, encoding oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am J Hum Genet 2015;97:848–54.
[Article] [PubMed] [PMC]26. Kataoka K, Kurokawa M. Ecotropic viral integration site 1, stem cell self-renewal and leukemogenesis. Cancer Sci 2012;103:1371–7.
[Article] [PubMed] [PMC]27. Perkins AS, Mercer JA, Jenkins NA, Copeland NG. Patterns of Evi-1 expression in embryonic and adult tissues suggest that Evi-1 plays an important regulatory role in mouse development. Development 1991;111:479–87.
[Article] [PubMed]28. Greenhalgh KL, Howell RT, Bottani A, Ancliff PJ, Brunner HG, Verschuuren-Bemelmans CC, et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet 2002;39:876–81.
[Article] [PubMed] [PMC]29. Kim YH, Yang JS, Lee YJ, Bae MH, Park KH, Lee DH, et al. 1q21.1 microdeletion identified by chromosomal microarray in a newborn with upper airway obstruction. J Genet Med 2018;15:34–7.
[Article]30. We JS, Park IY, Kim MS, Shin JC. Prenatal diagnosis of thrombocytopeniaabsent radius syndrome using three-dimensional ultrasound. Korean J Obstet Gynecol 2008;51:665–9.31. Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 2012;44:435–9. , S1-2.
[Article] [PubMed] [PMC]32. Gunay-Aygun M, Huizing M, Gahl WA. Molecular defects that affect platelet dense granules. Semin Thromb Hemost 2004;30:537–47.
[Article] [PubMed] [PMC]33. Chiang PW, Oiso N, Gautam R, Suzuki T, Swank RT, Spritz RA. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS4 proteins are components of two complexes, BLOC-3 and BLOC-4, involved in the biogenesis of lysosome-related organelles. J Biol Chem 2003;278:20332–7.
[Article] [PubMed]34. Witkop CJ, Nuñez Babcock M, Rao GH, Gaudier F, Summers CG, Shanahan F, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R 1990;82:333–9.
[PubMed]35. Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol 2008;15:22–9.
[Article] [PubMed]36. Ward DM, Shiflett SL, Kaplan J. Chediak-Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Curr Mol Med 2002;2:469–77.
[Article] [PubMed]37. Park GS, Lee DW, Song MY, Kim HK, Han KJ, Cho BK. Chediak-Higashi syndrome with hyperpigmentation. Ann Dermatol 1996;8:140–3.
[Article]38. Seri M, Cusano R, Gangarossa S, Caridi G, Bordo D, Lo Nigro C, et al. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Heggllin/Fechtner Syndrome Consortium. Nat Genet 2000;26:103–5.
[Article] [PubMed]39. Pecci A, Noris P, Invernizzi R, Savoia A, Seri M, Ghiggeri GM, et al. Immunocytochemistry for the heavy chain of the non-muscle myosin IIA as a diagnostic tool for MYH9-related disorders. Br J Haematol 2002;117:164–7.
[Article] [PubMed]40. Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, et al. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: MayHegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am J Hum Genet 2001;69:1033–45.
[Article] [PubMed] [PMC]41. Saito H, Kunishima S. Historical hematology: May-Hegglin anomaly. Am J Hematol 2008;83:304–6.
[Article] [PubMed]42. Seri M, Pecci A, Di Bari F, Cusano R, Savino M, Panza E, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore) 2003;82:20315
[Article]43. Han KH, Lee H, Kang HG, Moon KC, Lee JH, Park YS, et al. Renal manifestations of patients with MYH9-related disorders. Pediatr Nephrol 2011;26:549–55.
[Article] [PubMed]44. Jang MJ, Park HJ, Chong SY, Huh JY, Kim IH, Jang JH, et al. A Trp33Arg mutation at exon 1 of the MYH9 gene in a Korean patient with May-Hegglin anomaly. Yonsei Med J 2012;53:662–6.
[Article] [PubMed] [PMC]45. Kook H, Nam HS, Baek HJ, Kim YO, Eom GH, Kee HJ, et al. Clinical characteristics of autosomal dominant giant platelet syndromes and mutation analysis of MYH9. Korean J Hematol 2006;41:16–27.
[Article]46. Lee NH, Kim ES, Sung SI, Ahn SY, Lee MS, Han YM, et al. May-Hegglin anomaly diagnosed by genetic study in a newborn infant. Korean J Perinatol 2012;23:108–12.47. Oh T, Jung Seo H, Taek Lee K, Jo Kim H, Jun Kim H, Lee JH, et al. MYH9 nephropathy. Kidney Res Clin Pract 2015;34:53–6.
[Article] [PubMed]48. Park SJ, Wy H, Jung HL, Shim JW, Shim JY, Kim DS, et al. A case of myosin-heavy-chain-9 (MYH9) gene mutation confirmed May-Hegglin anomaly: 11-year Follow-up. Clin Pediatr Hematol Oncol 2016;23:16770
[Article]49. Rhim JW, Kim KH, Kim DS, Kim BS, Kim JS, Kim CH, et al. Prevalence of primary immunodeficiency in Korea. J Korean Med Sci 2012;27:788–93.
[Article] [PubMed] [PMC]50. Sabri S, Foudi A, Boukour S, Franc B, Charrier S, Jandrot-Perrus M, et al. Deficiency in the Wiskott-Aldrich protein induces premature proplatelet formation and platelet production in the bone marrow compartment. Blood 2006;108:134–40.
[Article] [PubMed]51. Zhu Q, Zhang M, Blaese RM, Derry JM, Junker A, Francke U, et al. The Wiskott-Aldrich syndrome and X-linked congenital thrombocytopenia are caused by mutations of the same gene. Blood 1995;86:3797–804.
[Article] [PubMed]52. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 1994;125(6 Pt 1): 87685
[Article]53. Jo EK, Futatani T, Kanegane H, Kubota T, Lee YH, Jung JA, et al. Mutational analysis of the WASP gene in 2 Korean families with WiskottAldrich syndrome. Int J Hematol 2003;78:40–4.
[Article] [PubMed]54. Kang HJ, Shin HY, Ko SH, Park JA, Kim EK, Rhim JW, et al. Unrelated bone marrow transplantation with a reduced toxicity myeloablative conditioning regimen in Wiskott-Aldrich syndrome. J Korean Med Sci 2008;23:146–8.
[Article] [PubMed] [PMC]55. Kim HJ, Yoo EH, Ki CS, Yoo GH, Koo HH, Kim JW, et al. A novel mutation W252X in the WAS gene in a Korean patient with Wiskott-Aldrich syndrome. Int J Hematol 2006;83:426–8.
[Article] [PubMed]56. Kim MK, Kim ES, Kim DS, Choi IH, Moon T, Yoon CN, et al. Two novel mutations of Wiskott-Aldrich syndrome: the molecular prediction of interaction between the mutated WASP L101P with WASP-interacting protein by molecular modeling. Biochim Biophys Acta 2004;1690:13440
[Article]57. Lee EK, Eem YJ, Chung NG, Kim MS, Jeong DC. A case of familial X-linked thrombocytopenia with a novel WAS gene mutation. Korean J Pediatr 2013;56:265–8.
[Article] [PubMed] [PMC]58. Lee YH, Lim YJ, Shin SA, Song CH, Jo EK, Jung JA, et al. Phenotypic and genotypic correction of WASP gene mutation in Wiskott-Aldrich syndrome by unrelated cord blood stem cell transplantation. J Korean Med Sci 2009;24:751–4.
[Article] [PubMed] [PMC]59. Park SK, Kim CS, Song DK, Kim JY, Choi IJ, Kim DK. A familial case of Wiskott-Aldrich Syndrome with a hotspot mutation in exon 2 of the WAS Gene. J Korean Med Sci 2007;22:998–1001.
[Article] [PubMed] [PMC]60. Yoon SH, Cho T, Kim HJ, Kim SY, Ko JH, Baek HS, et al. IVS6+5G>A found in Wiskott-Aldrich syndrome and X-linked thrombocytopenia in a Korean family. Pediatr Blood Cancer 2012;58:297–9.
[Article] [PubMed]61. Yi ES, Choi YB, Lee NH, Lee JW, Sung KW, Koo HH, et al. Allogeneic hematopoietic cell transplantation in patients with primary immunodeficiencies in Korea: eleven-year experience in a single center. J Clin Immunol 2018;38:757–66.
[Article] [PubMed]62. Afrasiabi A, Artoni A, Karimi M, Peyvandi F, Ashouri E, Mannucci PM. Glanzmann thrombasthenia and Bernard-Soulier syndrome in south Iran. Clin Lab Haematol 2005;27:324–7.
[Article] [PubMed]63. Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood 2011;118:5996–6005.
[Article] [PubMed]64. Fang J, Nurden P, North P, Nurden AT, Du LM, Valentin N, et al. C560Rβ3 caused platelet integrin αII b β3 to bind fibrinogen continuously, but resulted in a severe bleeding syndrome and increased murine mortality. J Thromb Haemost 2013;11:1163–71.
[Article] [PubMed] [PMC]65. Park KJ, Chung HS, Lee KO, Park IA, Kim SH, Kim HJ. Novel and recurrent mutations of ITGA2B and ITGB3 genes in Korean patients with Glanzmann thrombasthenia. Pediatr Blood Cancer 2012;59:335–8.
[Article] [PubMed]66. George JN, Caen JP, Nurden AT. Glanzmann's thrombasthenia: the spectrum of clinical disease. Blood 1990;75:1383–95.
[Article] [PubMed]67. Wagner CL, Mascelli MA, Neblock DS, Weisman HF, Coller BS, Jordan RE. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood 1996;88:907–14.
[Article] [PubMed]68. Savoia A, Kunishima S, De Rocco D, Zieger B, Rand ML, Pujol-Moix N, et al. Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat 2014;35:1033–45.
[Article] [PubMed]69. López JA, Andrews RK, Afshar-Kharghan V, Berndt MC. Bernard-Soulier syndrome. Blood 1998;91:4397–418.
[Article] [PubMed]70. Poujol C, Ware J, Nieswandt B, Nurden AT, Nurden P. Absence of GPIbalpha is responsible for aberrant membrane development during megakaryocyte maturation: ultrastructural study using a transgenic model. Exp Hematol 2002;30:352–60.
[Article] [PubMed]71. Berndt MC, Shen Y, Dopheide SM, Gardiner EE, Andrews RK. The vascular biology of the glycoprotein Ib-IX-V complex. Thromb Haemost 2001;86:178–88.
[Article] [PubMed]72. Pham A, Wang J. Bernard-Soulier syndrome: an inherited platelet disorder. Arch Pathol Lab Med 2007;131:1834–6.
[Article] [PubMed]73. Miller JL, Cunningham D, Lyle VA, Finch CN. Mutation in the gene encoding the alpha chain of platelet glycoprotein Ib in platelet-type von Willebrand disease. Proc Natl Acad Sci U S A 1991;88:4761–5.
[Article] [PubMed] [PMC]74. Russell SD, Roth GJ. Pseudo-von Willebrand disease: a mutation in the platelet glycoprotein Ib alpha gene associated with a hyperactive surface receptor. Blood 1993;81:1787–91.
[Article] [PubMed]75. Randi AM, Rabinowitz I, Mancuso DJ, Mannucci PM, Sadler JE. Molecular basis of von Willebrand disease type IIB. Candidate mutations cluster in one disulfide loop between proposed platelet glycoprotein Ib binding sequences. J Clin Invest 1991;87:1220–6.
[Article] [PubMed] [PMC]76. Budarf ML, Konkle BA, Ludlow LB, Michaud D, Li M, Yamashiro DJ, et al. Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. Hum Mol Genet 1995;4:763–6.
[Article] [PubMed]77. Burnside RD. 22q11.21 Deletion syndromes: a review of proximal, central, and distal deletions and their associated features. Cytogenet Genome Res 2015;146:89–99.
[Article] [PubMed]78. Nakagawa M, Okuno M, Okamoto N, Fujino H, Kato H. Bernard-Soulier syndrome associated with 22q11.2 microdeletion. Am J Med Genet 2001;99:286–8.
[Article] [PubMed]79. Johnson B, Doak R, Allsup D, Astwood E, Evans G, Grimley C, et al. A comprehensive targeted next-generation sequencing panel for genetic diagnosis of patients with suspected inherited thrombocytopenia. Res Pract Thromb Haemost 2018;2:640–52.
[Article] [PubMed] [PMC]80. Sánchez-Guiu I, Antón AI, Padilla J, Velasco F, Lucia JF, Lozano M, et al. Functional and molecular characterization of inherited platelet disorders in the Iberian Peninsula: results from a collaborative study. Orphanet J Rare Dis 2014;9:213
[Article] [PubMed] [PMC]81. Schlemper RJ, van der Maas AP, Eikenboom JC. Familial essential thrombocythemia: clinical characteristics of 11 cases in one family. Ann Hematol 1994;68:153–8.
[Article] [PubMed]82. Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood 2004;103:4198–200.
[Article] [PubMed]83. Paterson AD, Rommens JM, Bharaj B, Blavignac J, Wong I, Diamandis M, et al. Persons with Quebec platelet disorder have a tandem duplication of PLAU, the urokinase plasminogen activator gene. Blood 2010;115:12646
[Article]84. Kahr WH, Hinckley J, Li L, Schwertz H, Christensen H, Rowley JW, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet 2011;43:738–40.
[Article] [PubMed] [PMC]85. Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001;409:202–7.
[Article] [PubMed]86. Hirata T, Kakizuka A, Ushikubi F, Fuse I, Okuma M, Narumiya S. Arg60 to Leu mutation of the human thromboxane A2 receptor in a dominantly inherited bleeding disorder. J Clin Invest 1994;94:1662–7.
[Article] [PubMed] [PMC]87. Mestel F, Oetliker O, Beck E, Felix R, Imbach P, Wagner HP. Severe bleeding associated with defective thromboxane synthetase. Lancet 1980;1:157
[Article]88. Geneviève D, Proulle V, Isidor B, Bellais S, Serre V, Djouadi F, et al. Thromboxane synthase mutations in an increased bone density disorder (Ghosal syndrome). Nat Genet 2008;40:284–6.
[Article] [PubMed]89. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010;468:834–8.
[Article] [PubMed]90. Hwang DJ, Yang JW, Kim SY, Yi HK, Lee DY, Hwang PH. Diagnostic approach of Wiskott-Aldrich syndrome. Korean J Pediatr 2004;47:7263491. Baek HJ, Choi SH, Sohn KR, Kook H, Kim SJ, Song ES, et al. Mutaion analysis in X-linked recessive congenital immunodeficiency syndromes. Chonnam Med J 2005;41:48–61.