About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 57(1); 2014
Identification of a novel mutation in the CHD7 gene in a patient with CHARGE syndrome
- Corresponding author: Jihyun Kim, MD. Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea. Tel: +82-2-3410-1035, Fax: +82-2-3410-0805, jhlovechild@gmail.com
- Received September 11, 2012 Revised October 10, 2012 Accepted October 16, 2012
- Abstract
-
CHARGE syndrome has been estimated to occur in 1:10,000 births worldwide and shows various clinical manifestations. It is a genetic disorder characterized by a specific and a recognizable pattern of anomalies. The major clinical features are ocular coloboma, heart malformations, atresia of the choanae, growth retardation, genital hypoplasia, and ear abnormalities. The chromodomain helicase DNA-binding protein 7 (CHD7) gene, located on chromosome 8q12.1, causes CHARGE syndrome. The CHD7 protein is an adenosine triphosphate (ATP)-dependent chromatin remodeling protein. A total of 67% of patients clinically diagnosed with CHARGE syndrome have CHD7 mutations. Five hundred twenty-eight pathogenic and unique CHD7 alterations have been identified so far. We describe a patient with a CHARGE syndrome diagnosis who carried a novel de novo mutation, a c.3896T>C (p. leu1299Pro) missense mutation, in the CHD7 gene. This finding will provide more information for genetic counseling and expand our understanding of the pathogenesis and development of CHARGE syndrome.
- Introduction
- Introduction
CHARGE syndrome is heterogenous collection of congenital anomalies. The major clinical features are ocular coloboma, heart malformations, atresia of the choanae, growth retardation, genital hypoplasia, and ear abnormalities. Major features are coloboma, atresia of the choanae, and hypoplastic semicircular canals. Minor features are rhombencephalic dysfunction, hypothalamo-hypophyseal dysfunction, abnormal middle or external ears, malformation of mediastinal organs, and mental retardation. Typical CHARGE syndrome refers to three major features or two major and two minor criteria. Partial or incomplete CHARGE syndrome is defined as two major and one minor criteria1). Other clinical findings such as dysmorphic features, cleft lip/palate, and arhinencephaly can appear2). Hearing loss and hypoplasia of the semicircular canals have been recently proposed as major CHARGE syndrome diagnostic criteria, because almost all patients have these symptoms3).The chromodomain helicase DNA-binding protein 7 (CHD7) gene, located on chromosome 8q12.1, causes CHARGE syndrome. A total of 67% of patients clinically diagnosed with CHARGE syndrome have CHD7 mutations4). The CHD7 gene encodes a protein that participates in chromatin organization and plays a predominant role in neural tissue during fetal development5). Most of the identified mutations are scattered throughout the gene, showing no preferential domain. The majority of CHD7 gene mutations occurs de novo. Familial transmission and germline mosaicism have rarely been identified.We report here a case of typical CHARGE syndrome carrying a novel de novo mutation in the gene encoding CHD7.
- Case report
- Case report
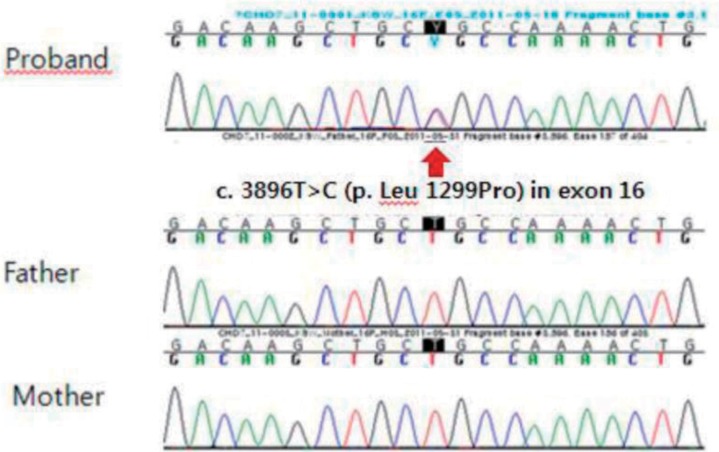
A 21 day-old girl was transferred to Samsung Medical Center due to extubation failure. The baby was born at the 35+1 weeks of gestation by emergency cesarean section due to spontaneous rupture of the membranes and premature labor. Apgar scores were 2 at 1 minute and 8 at 5 minutes. When the baby was born, she did not cry and became cyanotic and lethargic. Positive pressure ventilation was not effective, so endotracheal intubation was performed in the delivery room. At birth, weight, length, and head circumference were 2,030 g (25th percentile), 49 cm (75th-90th percentile), and 35 cm (>90th percentile), and she had low set ears. A large patent ductus arteriosus (PDA) was found and ligated 12 days after birth.We conducted osteomeatal unit computed tomography (CT) to find the cause of extubation failure. CT scan showed bilateral choanal atresia, atresia of the left external auditory canal, and stenosis of the right auditory canal (Fig. 1). Thirty-five days after birth, she underwent a repair operation of the choanae, and we tried to extubate again. However, she was reintubated with an endotracheal tube because of sustained desaturation and tachypnea. We considered that the cause of extubation failure was an upper airway problem. She was identified to have postglottic stenosis through laryngomicrosurgery (Fig. 2). Forty-two days after birth, a tracheostomy was performed, and respiration stabilized without respiratory support.The patient had a left aortic arch with aberrant origin of the right subclavian artery on chest CT and minimal left pulmonary artery hypoplasia without focal stenosis on echocardiography. Based on these results, we conducted the ophthalmic examination to identify other features of CHARGE syndrome, and found coloboma in her both eyes. Because she met two out of the three major criteria and two of the five minor diagnostic criteria, we concluded that she had typical CHARGE syndrome.Written informed consent was obtained for a molecular genetic analysis of the CHD7 gene mutation from the parents. Direct sequencing analysis of the CHD7 gene was performed with genomic DNA isolated from peripheral blood leukocytes. As a result, a c.3896T>C (p. leu1299Pro) missense mutation in exon 16 of the CHD7 gene was detected, which has not been reported previously (Fig. 3). A familial study revealed that neither of the parents had the mutation. The polymorphism phenotyping program and the sorting intolerant from tolerant program predicted "possibly damaging" and "not tolerated." The amino acid residue L1299 is highly conserved across different species, and the L1299P variation does not occur in 200 control chromosomes from 100 healthy individuals.After discharge at 2 months of age, she has been followed up at the outpatient clinic regularly. At present, she is 11 months old and is using a hearing aid because of sensorineural hearing loss. Since she has had recurrent aspiration; thus G-tube feeding is required. Her height was 10th-25th percentile, body weight was 5th-10th percentile, and head circumference was 10th-25th percentile. The patient can sit alone but cannot stand with support. Hypoplasia of both kidneys was identified by abdominal ultrasonography, which was performed at 5 months of age. She has met two of the three major criteria and four of the five minor diagnostic criteria for CHARGE syndrome.
- Discussion
- Discussion
CHARGE syndrome has been estimated to occur in 1:10,000 births worldwide and shows various clinical manifestations6). As diagnosis is primarily established by clinical symptoms, it is important to know usual clinical symptoms. Coloboma in patients with CHARGE syndrome usually involves bilateral choroid, retina, and the optic nerve. Typical colobama in CHARGE syndrome cause field defects7). Choanal atresia is considered a pivotal cause of respiratory difficulty during the neonatal period4). Heart defects are present in 75%-85% of patients with CHARGE syndrome, and common heart defects are tetralogy of Fallot, PDA, and atrial septal defects7). A double outlet right ventricle and hypoplastic left/right heart are rarely reported. Hearing loss can be difficult to identify in a pediatric patient, as it requires multiple brain stem audio evoked response tests over several months.Children with CHARGE syndrome usually have normal birth weights and lengths. However, linear growth usually declines in late infancy. It has been suggested that pituitary hormone deficiencies of growth hormone and thyroid stimulating hormone cause the growth defects7). Therefore, patients with CHARGE syndrome must have their growth monitored. Additionally, hypogonadotropic hypogonadism and delayed puberty can occur8).Neonates with CHARGE syndrome often have multiple life-threatening medical conditions related to respiratory difficulty, as in our case. Studies have shown that 15%-60% of patients with CHARGE syndrome require tracheostomy9,10). Blake et al.11) reported a poor prognosis if patients have cyanotic cardiac lesions, bilateral posterior choanal atresia, and a tracheoesophageal fistula. Feeding difficulties also are a major cause of morbidity. Therefore, neonates with suspected CHARGE syndrome should have eye, heart, airway, and swallowing dysfunctions checked.With regard to the pathogenesis, CHD7 gene is known to be responsible for the development of CHARGE syndrome. The CHD7 protein is an ATP-dependent chromatin remodeling protein. It consists of several important domains that play a role in transcription activation and repression during chromatin remodeling. Although the function of this protein is unclear, a role in controlling gene expression programs in embryonic stem cells and other cell has been suggested5). Identifying CHD7 protein function is important to understand the pathogenesis of CHARGE syndrome.In that sense, CHD7 molecular testing confirms the diagnosis in the majority of cases. As of June 2011, 528 CHD7 alterations have been identified in 802 index patients with CHARGE syndrome12). A sequence analysis is the first diagnostic tool, because most mutations may include small intragenic deletions/insertions and missense, nonsense, and splice site mutations. If a mutation is not identified by sequence analysis, a deletion/duplication analysis can be performed to identify large gene deletions. Although almost patients with CHARGE syndrome have a normal karyotype, chromosomal abnormalities that disrupt CHD7 have been rarely reported; balanced chromosomal translocation t(6;8)(6p8p;6q8q)13), de novo balanced chromosomal rearrangement t(8;13)(q11.2;q22)14), and interstitial deletion of 8q11.2-q1315). Therefore, a cytogenetic analysis can be used to exclude chromosomal abnormalities and other syndromes overlapping CHARGE syndrome.In summary, we identified a novel CHD7 gene mutation in a girl with CHARGE syndrome. This finding will provide more information for genetic counseling and expand our understanding of the pathogenesis and development of CHARGE syndrome.
- Conflicts of interest
No potential conflict of interest relevant to this article was reported.
- References
- 1. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A 2005;133A:306–308.
[Article] [PubMed]2. Wessels K, Bohnhorst B, Luhmer I, Morlot S, Bohring A, Jonasson J, et al. Novel CHD7 mutations contributing to the mutation spectrum in patients with CHARGE syndrome. Eur J Med Genet 2010;53:280–285.
[Article] [PubMed]3. Lee YW, Kim SC, Shin YL, Kim JW, Hong HS, Lee YK, et al. Clinical and genetic analysis of the CHD7 gene in Korean patients with CHARGE syndrome. Clin Genet 2009;75:290–293.
[Article] [PubMed]4. Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr 2006;148:410–414.
[Article] [PubMed]5. Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet 2006;43:211–217.
[Article] [PubMed]6. Issekutz KA, Graham JM Jr, Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A 2005;133A:309–317.
[Article] [PubMed]7. Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet 2006;78:303–314.
[Article] [PubMed]8. Dauber A, Hirschhorn JN, Picker J, Maher TA, Milunsky A. Delayed puberty due to a novel mutation in CHD7 causing CHARGE syndrome. Pediatrics 2010;126:e1594–e1598.
[Article] [PubMed]9. Roger G, Morisseau-Durand MP, Van Den Abbeele T, Nicollas R, Triglia JM, Narcy P, et al. The CHARGE association: the role of tracheotomy. Arch Otolaryngol Head Neck Surg 1999;125:33–38.
[Article] [PubMed]10. White DR, Giambra BK, Hopkin RJ, Daines CL, Rutter MJ. Aspiration in children with CHARGE syndrome. Int J Pediatr Otorhinolaryngol 2005;69:1205–1209.
[Article] [PubMed]11. Blake KD, Russell-Eggitt IM, Morgan DW, Ratcliffe JM, Wyse RK. Who's in CHARGE? Multidisciplinary management of patients with CHARGE association. Arch Dis Child 1990;65:217–223.
[Article] [PubMed] [PMC]12. Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat 2012;33:1149–1160.
[Article] [PubMed]13. Hurst JA, Meinecke P, Baraitser M. Balanced t(6;8)(6p8p;6q8q) and the CHARGE association. J Med Genet 1991;28:54–55.
[Article] [PubMed] [PMC]