About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 56(2); 2013
Cardiomyopathies in children
- Corresponding author: Young Mi Hong, MD. Department of Pediatrics, Ewha Womans University School of Medicine, 1071 Anyangcheon-ro, Yangcheon-gu, Seoul 158-710, Korea. Tel: +82-2-2650-2841, Fax: +82-2-2653-3718, hongym@chollian.net
- Received September 21, 2012 Accepted January 04, 2013
- Abstract
-
Cardiomyopathy (CMP) is a heterogeneous disease caused by a functional abnormality of the cardiac muscle. CMP is of 2 major types, dilated and hypertrophic, and is further classified as either primary or secondary. Secondary CMP is caused by extrinsic factors, including infection, ischemia, hypertension, and metabolic disorders. Primary CMP is diagnosed when the extrinsic factors of secondary CMP are absent. Furthermore, the World Health Organization, American Heart Association, and European Cardiology Association have different systems for clinically classifying primary CMP. Primary CMP is rare and associated with a family history of the disease, implying that genetic factors might affect its incidence. In addition, the incidence of CMP varies widely according to patient ethnicity. Genetic testing plays an important role in the care of patients with CMP and their families because it confirms diagnosis, determines the appropriate care for the patient, and possibly affects patient prognosis. The diagnosis and genetic identification of CMP in patients' families allow the possibility to identify novel genes that may lead to new treatments. This review focuses on the epidemiology, pathophysiology, diagnosis, and treatment of CMP, with the aim of providing pediatricians with insights that may be helpful in the early identification and management of idiopathic CMP in children.
- Introduction
- Introduction
An understanding of cardiomyopathy (CMP) is very important, as it is a common cause of heart failure in children and the most common indication for heart transplantation in children older than 1 year1).Primary CMP is rare2-4). In Asia, reports on the incidence of primary CMP in pediatric age groups are extremely rare; hence, further research is needed in this area. Among Asian countries, Korea is uniquely comprised of a relatively homogeneous ethnic group; therefore, a study of the incidence of CMP in Korean children would be especially interesting4).Dilated CMP (DCMP) is the most common form of CMP worldwide and has many causes. In 30% to 48% of patients, DCMP is genetically inherited. Moreover, inflammatory disorders such as myocarditis, or toxic agents such as medications and alcohol can result in DCMP5). Of all the DCMP cases, 20% to 48% have a family history of the disease6).The etiology of hypertrophic CMP (HCMP) in the pediatric population is heterogeneous, including inborn metabolism errors, neuromuscular disorders, and malformation syndromes. However, most cases of apparently idiopathic HCMP in childhood are caused by mutations of cardiac sarcomere protein genes.In the pediatric population, risk stratification is necessary because most cases of HCMP have a family history of the disease. Evaluation of first-degree relatives and any at-risk family members should be a routine component of clinical management7).Recent technological developments in genetic studies on CMP have elucidated the potential advantage of genetic testing in the diagnosis and in understanding the pathogenetic basis of inherited CMP7).
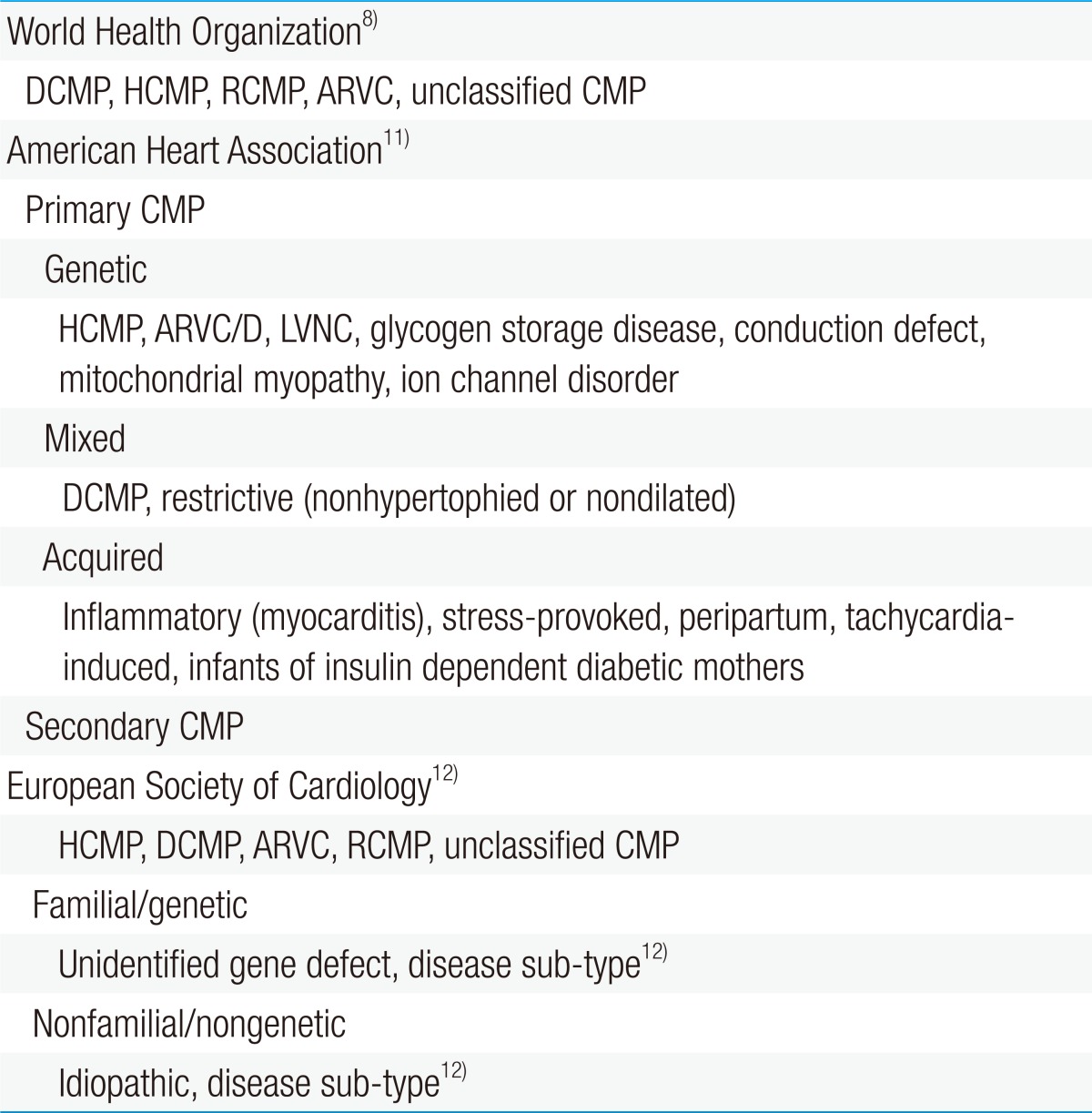
- Classification of CMP
- Classification of CMP
The World Health Organization (WHO) defines CMP as "a disease of the myocardium associated with cardiac dysfunction" and classifies it as dilated, hypertrophic, restrictive, arrhythmogenic right ventricular, or unclassified (Table 1)8).Some consider this definition by the WHO as inadequate to emphasize the complexity of the disease9). Therefore, the American Heart Association (AHA) issued a consensus statement that revised the WHO's classification of CMP based on genetics, heart structural changes, cellular events, and multiorgan involvement10).Most classify CMP as either primary or secondary. CMP can be considered as primary when the disorder is solely or predominantly confined to the heart muscle and has a genetic, nongenetic, or acquired etiology. Meanwhile, secondary CMP is diagnosed when the disorder involves myocardial damage because of a systemic or multiorgan disease11).The European Society of Cardiology has adopted an alternative classification which is similar to the WHO definition in some ways but is more clinically orientated12).Each of the CMP forms is determined by the nature of muscle damage. Moreover, CMP may be classified as more than 1 type or may change from 1 type to another over time in some patients. Although the WHO has not formally categorized left ventricular noncompaction cardiomyopathy, it is increasingly being identified8).
- Incidence
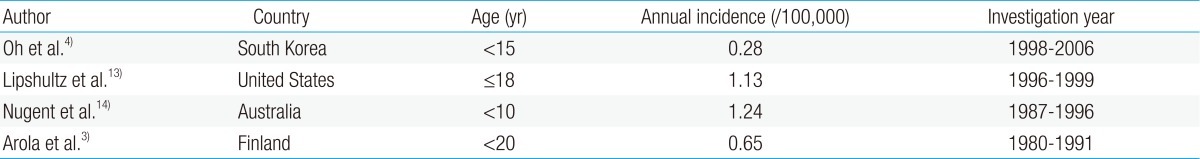
- Incidence
Reports on the incidence of CMP in children are rare. The incidence of CMP in different countries has been inconsistent owing to the varying research methods used in studies with different age groups and study durations (Table 2).According to a study based on the AHA classification, the incidence of CMP between 1998 and 2006 in Korean children was 0.28 per 100,000 children younger than 15 years4). The point prevalence of primary CMP in Korean children was 2.11 per 100,000 children on December 31, 2006. DCMP, HCMP, restrictive cardiomyopathy (RCMP), and others accounted for 66.4%, 23.5%, 6.5%, and 3.6%, respectively4).According to the United States (US) Pediatric Cardiomyopathy Registry, the annual incidence of CMP was 1.13 per 100,000 children younger than 18 years13), with DCMP as the most common (58%), followed by HCMP (30%). There were relatively few cases of RCMP (5%) and arrhythmogenic right ventricular cardiomyopathy (ARVC) (5%).The incidence of CMP in children was 1.24 per 100,000 children younger than 10 years in Australia14) and 0.65 per 100,000 children aged 20 years or younger in Finland3).The incidence of DCMP in Korean children was 0.18 per 100,000 children4), whereas that in American children was 0.57 per 100,000 children younger than 18 years15). These findings suggest that the incidence of primary CMP is significantly lower in Korean children than in other ethnic groups.The incidence of HCMP in Korean children was 0.07 per 100,000 children. Epidemiological studies in Finland, Australia, and the US revealed that the incidence of new cases of HCMP was between 0.24 and 0.47 per 100,000 children3,14,15).The differences in the incidence of HCMP between these studies may reflect the diversity in racial composition and dynamic fluxes in the study population, the inclusion of secondary cases of HCMP, the influence of family cascade screening, the age group under study, and the inclusion of cases identified at the time of autopsy. For example, a Korean study on HCMP that included only patients younger than 15 years as subjects might have excluded a larger proportion of HCMP patients because HCMP usually presents during the late adolescent years.Further studies with a more consistent approach in data collection and an international agreement on a unified classification system of CMP are needed.
- Causes
- Causes
- 1. Familial or genetic causes
- 1. Familial or genetic causes
Most CMP patients have a family history of the disease; hence, CMP is probably genetically transmitted. The genetic components are possibly mutations in the DNA spiral, the protein structure of many genes5,7).Current research is being conducted to identify the specific genes that cause CMP and to better understand how these genetic abnormalities contribute to the disease. However, CMP is a complex disease, with multiple diverse genes producing extremely variable outcomes7).Many children with HCMP (50% to 60%) or DCMP (30% to 48%) have a family history of the disease16). Recent genetic research has indicated that HCMP involves defects in the sarcomere genes and that it can be inherited in an autosomal dominant manner. However, DCMP involves defects in the cytoskeleton genes and can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner15).- 2. Acquired causes
- 2. Acquired causes
The most common cause of acquired CMP is myocarditis5). Other causes of acquired CMP include (1) cardiovascular conditions (e.g., Kawasaki disease, hypertension, congenital heart defect, cardiac transplantation, or surgery), (2) inflammatory or infectious diseases, (3) immunological diseases (e.g., human immunodeficiency virus), (4) toxin reactions (e.g., drug, alcohol, or radiation exposure), (5) obesity or dietary deficiencies, (6) connective tissue and autoimmune diseases, and (7) endocrine diseases. Persistent heart rhythm problems or problems of the coronary arteries, either congenital or acquired, can also lead to weakening of the heart5,7).Of the many causes of CMP, only few are directly treatable; therefore, most therapies are aimed at treating the secondary effects on the heart.According to the US Pediatric Cardiomyopathy Registry13), CMP can be categorized based on the following specific genetic causes of the disease: (1) myocarditis and other viral infections (27%), (2) familial inherited CMPs (24%), (3) neuromuscular disorders associated with CMP (22%), (4) metabolic disorders (16%), and (5) malformation syndromes associated with CMP (10%).- 3. Neuromuscular diseases
- 3. Neuromuscular diseases
Neuromuscular diseases associated with CMP include those that affect the nerves or skeletal muscles. These include muscular dystrophies (e.g., Duchenne and Becker muscular dystrophy), congenital myopathies, metabolic myopathies, and ataxias (e.g., Friedreich ataxia). Almost all of the neuromuscular diseases associated with CMP have a genetic basis5,7).- 4. Metabolic disorders
- 4. Metabolic disorders
Inborn errors of metabolism result in numerous infiltrative storage diseases, abnormal energy production, biochemical deficiencies, and disorders related to toxic substances accumulating in the heart. This category also includes mitochondrial abnormalities (e.g., respiratory chain diseases or mitochondrial myopathies), Pompe disease, Barth syndrome, and fatty acid oxidation defects (carnitine deficiency)5,7).- 5. Malformation syndrome
- 5. Malformation syndrome
Minor and major physical abnormalities with distinctive facial features are common in malformation syndrome cases. Malformation syndrome is caused by genetic mutations through autosomal dominant, autosomal recessive, or X-linked recessive inheritance. It can also be caused by a chromosomal defect where a specific chromosome is deleted or duplicated. The Noonan syndrome is the most common malformation syndrome associated with pediatric CMP5,7).- 6. Idiopathic CMP
- 6. Idiopathic CMP
In patients without any remarkable family history, the specific cause of heart muscle damage is yet to be known. Research is being conducted to classify the remaining "unknown cases" and determine whether any common genetic abnormalities are involved.
- Signs and Symptoms
- Signs and Symptoms
- 1. Dilated cardiomyopathy
- 1. Dilated cardiomyopathy
Infants and children with DCMP generally present with signs of congestive heart failure. Tachypnea, labored breathing, poor appetite, and slow weight gain are common symptoms in infants. In older children, there may be signs of poor exercise tolerance and gastrointestinal distress. In more severe cases, patients may experience syncope, arrhythmias, or sudden cardiac death5).Another sign of DCMP is greater dilation and a more spherical appearance of the left ventricle than usual, with raised wall stress and depressed systolic function. In addition, mitral regurgitation and ventricular arrhythmias can also develop. In the most severe cases, affected individuals present with symptoms and signs of heart failure5).- 2. Hypertrophic cardiomyopathy
- 2. Hypertrophic cardiomyopathy
HCMP has a wide range of clinical presentations, from few or even no symptoms to serious complications, including heart failure and sudden death.HCMP is considered to be the most common structural cause of sudden cardiac death in individuals younger than 35 years, including competitive athletes. Patients with HCMP can experience various symptoms, including chest pain, symptoms related to pulmonary congestion, dyspnea, impaired consciousness such as syncopal and presyncopal episodes, and palpitations20).In the later stages of HCMP, symptoms similar to DCMP may present, such as coughing, abdominal pain, weakness, breathing difficulties, and fluid accumulation in the lungs and body.According to a Korean registry4), the symptoms of DCMP at the time of diagnosis include dyspnea, coughing, feeding difficulty, dyspnea on exertion (DOE), poor weight gain, and syncope; those of HCMP include dyspnea, coughing, feeding difficulty, DOE, poor weight gain, and syncope; and those of RCMP include dyspnea, coughing, feeding difficulty, DOE, and poor weight gain.Asymptomatic patients with DCMP showed initial signs of cardiomegaly, tachypnea, heart murmur, arrhythmia, and cyanosis; those with HCMP presented with cardiomegaly, tachypnea, heart murmur, arrhythmia, and cyanosis; and those with RCMP presented with cardiomegaly, tachypnea, heart murmur, arrhythmia, and cyanosis4).
The symptoms of CMP may differ in every child. Symptoms can be absent, mild, or severe. Heart murmurs are not always present in children with CMP. Only approximately one-third to one-half of HCMP patients experience heart murmurs, which are usually because of ventricular obstruction or leaking in 1 of the heart valves. Because CMP is not easily detected on physical examination, it is often diagnosed in its later stages19).
- Diagnosis
- Diagnosis
- 1. Noninvasive procedures
- 1. Noninvasive procedures
1) Chest radiography
1) Chest radiography
Chest radiographs often reveal cardiomegaly and increased pulmonary vascular markings that are consistent with pulmonary edema5).2) Electrocardiography
2) Electrocardiography
An electrocardiogram (ECG) can reveal sinus tachycardia, ST-T wave changes, Q waves, conduction disease, bundle-branch block, left ventricular hypertrophy (LVH), and ectopy, including supraventricular tachycardia, atrial fibrillation, and ventricular arrhythmias in DCMP patients20-22). Abnormal ECG findings are present in 75% to 95% of patients with HCMP20,21).3) Echocardiography
3) Echocardiography
Echocardiography is the most informative noninvasive test for diagnosing and classifying CMP, and determining the degree of dysfunction in the heart muscle.The diagnosis of DCMP is based on the presence of left ventricular enlargement and reduced systolic dysfunction with an ejection fraction <50% or, more stringently, <45%. Echocardiographic findings include left ventricular dilation and systolic dysfunction, with or without mitral regurgitation. In addition, pericardial effusion, especially in myocarditis and heart rhythm irregularities, can be observed5).Echocardiography in HCMP remains the gold standard for the diagnosis of HCMP in children. Left ventricular (LV) wall thickness >2 standard deviations above the body surface area-corrected mean (z score) is sufficient to make a diagnosis. Hypertrophy is defined as >15 mm in adults and >2 z-scores in children. Traditionally, the pattern of hypertrophy is asymmetric, and it affects more than 1 cardiac segment, preferentially affecting the anterior interventricular septum20, 23, 24). Echocardiography is useful in assessing the pattern of hypertrophy, global systolic function, diastolic performance, presence and mechanism of outflow tract obstruction, atrial dimensions, and valvular function7).LV outflow tract obstruction is present at rest in 25% to 40% of children with HCMP25,26). Early echocardiographic findings of abnormal diastolic function indices, including reduced early diastolic septal tissue Doppler velocity, prolonged isovolumic relaxation times, elevated septal transmitral E/Ea ratio, and increased left atrial volume, may precede the onset of LVH in children and adolescents27-29).4) Magnetic resonance imaging (MRI)
4) Magnetic resonance imaging (MRI)
Recently, MRI is playing an increasing number of roles in the management of patients with HCMP7). MRI has been shown to be more effective in imaging difficult to visualize LV segments, including the LV apex and anterolateral free wall29,30). MRI has been shown to more accurately predict the degree of LVH than other procedures30,31).5) Genetic studies
5) Genetic studies
Because CMP can be a symptom of a genetic disorder (e.g., mitochondrial disorder, fatty acid oxidation disorder, and glycogen storage diseases), certain biochemical, genetic, and enzyme deficiency tests should be performed before determining the most appropriate medical therapy5).Genes that cause DCMP generally encode cytoskeletal and sarcomeric proteins, although the disturbance of calcium homeostasis also appears to be important. However, the causal roles of disrupted mitochondrial function and metabolic abnormalities in pediatric cases should be considered5).Familial HCMP is a genetically heterogeneous disorder, which means that a mutation in more than 1 gene can lead to the same condition. There are at least 13 causative genes that have been identified to date. They primarily encode sarcomere or sarcomere-related proteins. They include the cardiac-myosin heavy chain, myosin-binding protein C, cardiac troponin T, tropomyosin, cardiac troponin I, essential and regulatory myosin light chain, titin, and actinin-2 genes. A single mutation in any of these genes will lead to HCMP20). Most recently, multiple mutations have been identified in individuals who appear to develop clinically more severe diseases20).6) Biomarkers
6) Biomarkers
The B-type natriuretic peptide (BNP) and N-terminal probrain natriuretic peptide are the most widely used biomarkers. An increase in BNP levels is associated with reduced left ventricular systolic function, hypertrophy, raised filling pressures, and myocardial ischemia. In adults and children, BNP levels can be used to differentiate symptoms related to lung disease from those of heart failure32).
- 2. Invasive procedures
- 2. Invasive procedures
1) Radionuclide ventriculography
1) Radionuclide ventriculography
Recent studies have indicated that radionuclide ventriculography may be the best tool to diagnose ARVC. It is used to assess contraction and filling of the ventricles at rest and with exertion.2) Cardiac catheterization
2) Cardiac catheterization
Angiograms may be obtained to determine heart function and check for mitral regurgitation. In addition, endomyocardial biopsy sampling is another method of defining the cause of the disease33). The histological examination of the heart muscle is clinically useful to distinguish between disease processes that need alternative treatment strategies, such as storage diseases, malignancies, sarcoidosis, and hemochromatosis.Identification of the causative virus by performing polymerase chain reaction of its viral genome, using endomyocardial biopsy samples, has been useful in establishing the cause of acute myocarditis34).
Invasive tests are sometimes necessary, especially in ARVC cases. These tests require anesthesia and are performed to determine the severity of CMP.
- Treatment
- Treatment
- 1. Medical therapy
- 1. Medical therapy
1) Dilated cardiomyopathy
1) Dilated cardiomyopathy
Patients with DCMP may be given an angiotensin-converting enzyme (ACE) inhibitor or anticongestive medications such as captopril, enalapril, and spironolactone.ACE inhibitor therapy is recommended for nearly all children with asymptomatic left ventricular dysfunction35,36).Diuretics are recommended for children with heart failure to achieve euvolemia and minimize congestive symptoms. The addition of digoxin is recommended if the child remains symptomatic. Digoxin is specifically not recommended for children with asymptomatic left ventricular dysfunction35).2) Hypertrophic cardiomyopathy
2) Hypertrophic cardiomyopathy
- (1) Affected individuals
- (1) Affected individuals
Sudden death occurs most commonly in patients with HCMP either during or immediately after exercise. Death can also occur at rest20).The following treatment options are currently available for HCMP patients: lifestyle modifications such as avoiding competitive sports and use of pharmacological agents such as calcium channel blockers, beta-blockers, and diuretics.Dual-chamber pacing, septal myotomy-myectomy, and transcoronary alcohol septal ablation of the myocardium are prescribed to individuals with significant left ventricular outflow tract obstruction who are unresponsive to drug therapy.However, the most important advancement in the clinical management of HCMP is the use of implantable cardioverter defibrillator (ICD) therapy to prevent sudden death20). Recent studies have shown that ICD therapy is the most definitive form of therapy for patients who have a high risk of sudden death, easily surpassing empirically based preventative strategies previously used in HCMP, such as amiodarone and beta-blockers.- (2) Asymptomatic family members
- (2) Asymptomatic family members
It is strongly recommended that all first-degree relatives of a patient be clinically screened for HCMP. Minimally, this should include a physical examination by a cardiologist, ECG, and transthoracic echocardiography20).There is some evidence that beta-blockade in asymptomatic pediatric patients with HCMP is effective in reducing the risk of sudden unexpected death. These findings have influenced some centers to routinely use beta-blockers in all patients with HCMP, obtaining excellent results37,38). However, it is not clear whether the routine use of beta-blockade in asymptomatic patients with HCMP has any definitive benefit. Calcium channel blockers are frequently used as an adjunct therapy in symptomatic adults with HCMP38).Diuretics and digoxin are not usually used for HCMP patients with ventricular obstruction because these drugs can actually worsen the obstruction of blood flow out of the heart. Instead, beta-blockers and calcium channel blockers are frequently prescribed to patients with moderate to severe obstruction. Medications such as propranolol and verapamil may be given to reduce the outflow obstruction by slowing the heart rate and relaxing the heart. Antiarrhythmic medications such as amiodarone and disopyramide may be required to reduce the risk of sudden cardiac death.
- 2. Cardiac resynchronization therapy
- 2. Cardiac resynchronization therapy
Use of cardiac resynchronization therapy has expanded the therapeutic options for pediatric and adult patients with advanced heart failure and ventricular conduction delay. Cardiac resynchronization therapy improves mechanical synchrony, which in turn increases left ventricular filling time, decreases mitral regurgitation, and reduces septal dyskinesis, which is frequently reported in adults5).- 3. Device implantation
- 3. Device implantation
A pacemaker or defibrillator is used when drugs are not effective in alleviating obstruction or when dangerous arrhythmias need to be regulated. Sudden death accounts for 50% of all deaths in children with HCMP. Hence, defibrillators are often recommended for children with HCMP, RCMP, and ARVC who show evidence of arrhythmias5).- 4. Surgical options
- 4. Surgical options
1) Myectomy
1) Myectomy
There is no single proven surgical technique for DCMP in children. A septal myectomy is sometimes recommended for symptomatic children with obstruction associated with HCMP39). The purpose of this surgery is to reduce heart failure symptoms related to restricted blood flow from the ventricles or severe mitral regurgitation.Another surgical technique is the use of ventricular assist devices. The use of ventricular assist devices has been shown to significantly improve the survival of adults and children with end-stage DCMP who are awaiting heart transplantation40).2) Heart transplantation
2) Heart transplantation
Heart transplantation is required in some extreme cases. Currently, transplants are reserved for patients in the most critical condition, such as those needing inotropes and, most commonly, mechanical ventilatory and mechanical device support5).CMP is the leading indication for heart transplantation in children. Within the first year of diagnosis, approximately 20% of infants and children who have symptomatic CMP require transplantation. Heart transplantation can eliminate all the symptoms of heart failure and greatly improve the survival of patients.
- 5. Additional treatments
- 5. Additional treatments
The outcomes of patients with DCMP and heart failure have greatly improved. However, it has been somewhat problematic to improve the outcomes of patients with nonischemic disease. Therefore, research in gene therapy41), stem-cell therapy42), and targeted treatments are being conducted.Several studies have noted beneficial effects of stem-cell transplantation in patients who developed depressed left ventricular systolic dysfunction after myocardial infarction43).
- Long-term prognosis
- Long-term prognosis
The CMP type and disease stage should be considered to determine the overall long-term prognosis of pediatric patients. The 9-year survival rate was estimated to be 69.8% in DCMP, 90.3% in HCMP, 47.2% in RCMP, and 42.0% in other CMP patients with ion channelopathy and mitochondrial CMP according to the Korean registry.4) Echocardiography has important implications for prognosis. A high degree of LVH could indicate a worse prognosis44,45).
- Screening
- Screening
It is recommended that all first-degree relatives of a patient be screened because CMP can be inherited and can present without any signs or symptoms. It is also advisable to screen second-degree relatives. This is strongly recommended if there is a family history of sudden infant death or sudden cardiac arrest.Children with an affected family member but without symptoms should undergo echocardiography and ECG every 1 to 3 years before the age of 12 years and then more frequently from age 12 to 21 years7,20).Those who have a family history of CMP may be advised to continue screening at least every 5 years throughout their adult life20).The generally accepted practice is that immediate family members of patients with HCMP should undergo periodic screenings for HCMP, including echocardiography, ECG, history taking, and physical examination19). Whether all children need to be screened for HCMP before participating in competitive athletics is controversial. Nevertheless, the Bethesda guidelines recommend universal screening, including historical evaluation and physical examination, before participation in competitive athletics because of the indolent nature of the disease, heterogeneity of clinical presentation, and potential for sudden unexpected death during peak exercise46).
- Conclusion
- Conclusion
The identification of the causes of gene mutations in CMP is critical because presymptomatic interventions have been shown to prevent morbidity and mortality. Identification of the mutated genes has the potential to lead to an understanding of the disease, including its etiology, pathophysiology, and potential therapy46). Genetic counseling should be regarded as an essential part of the genetic testing process and is recommended for all patients with HCMP47). Furthermore, as already discussed, early diagnosis, treatment, and potential risk stratification may result in improved patient survival. However, further international studies with a more consistent data collection approach for determining disease prevalence and an international agreement on a unified classification system for CMP are needed.
- References
- 1. Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, Orav EJ, et al. The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail Clin 2010;6:401–413.
[Article] [PubMed] [PMC]2. Bilgic A, Ozbarlas N, Ozkutlu S, Ozer S, Ozme S. Cardiomyopathies in children. Clinical, epidemiological and prognostic evaluation. Jpn Heart J 1990;31:789–797.
[Article] [PubMed]3. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, et al. Epidemiology of idiopathic cardiomyopathies in children and adolescents: a nationwide study in Finland. Am J Epidemiol 1997;146:385–393.
[Article] [PubMed]4. Oh JH, Hong YM, Choi JY, Kim SJ, Jung JW, Sohn S, et al. Idiopathic cardiomyopathies in Korean children: 9-year Korean multicenter study. Circ J 2011;75:2228–2234.
[Article] [PubMed]6. Taylor MR, Carniel E, Mestroni L. Cardiomyopathy, familial dilated. Orphanet J Rare Dis 2006;1:27
[Article] [PubMed] [PMC]7. Moak JP, Kaski JP. Hypertrophic cardiomyopathy in children. Heart 2012;98:1044–1054.
[Article] [PubMed]8. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 1996;93:841–842.
[Article] [PubMed]9. Giles TD. New WHO/ISFC classification of cardiomyopathies: a task not completed. Circulation 1997;96:2081–2082.
[PubMed]10. Thiene G, Corrado D, Basso C. Cardiomyopathies: is it time for a molecular classification? Eur Heart J 2004;25:1772–1775.
[Article] [PubMed]11. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006;113:1807–1816.
[Article] [PubMed]12. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008;29:270–276.
[Article] [PubMed]13. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003;348:1647–1655.
[Article] [PubMed]14. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003;348:1639–1646.
[Article] [PubMed]15. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006;296:1867–1876.
[Article] [PubMed]17. Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 2001;104:557–567.
[Article] [PubMed]18. Keren A, Syrris P, McKenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med 2008;5:158–168.
[Article] [PubMed]20. Semsarian C. CSANZ Cardiac Genetics Diseases Council Writing Group. Guidelines for the diagnosis and management of hypertrophic cardiomyopathy. Heart Lung Circ 2011;20:688–690.
[Article] [PubMed]21. Montgomery JV, Harris KM, Casey SA, Zenovich AG, Maron BJ. Relation of electrocardiographic patterns to phenotypic expression and clinical outcome in hypertrophic cardiomyopathy. Am J Cardiol 2005;96:270–275.
[Article] [PubMed]22. Maron BJ, McKenna WJ, Elliott P, Spirito P, Frenneaux MP, Keren A, et al. Hypertrophic cardiomyopathy. JAMA 1999;282:2302–2303.
[Article] [PubMed]23. Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol 1995;26:1699–1708.
[Article] [PubMed]24. Maron BJ, Gottdiener JS, Epstein SE. Patterns and significance of distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy: a wide angle, two dimensional echocardiographic study of 125 patients. Am J Cardiol 1981;48:418–428.
[Article] [PubMed]25. Maron BJ. Hypertrophic cardiomyopathy in childhood. Pediatr Clin North Am 2004;51:1305–1346.
[Article] [PubMed]26. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation 2005;112:1332–1338.
[Article] [PubMed]27. Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009;2:314–321.
[Article] [PubMed] [PMC]28. McMahon CJ, Nagueh SF, Pignatelli RH, Denfield SW, Dreyer WJ, Price JF, et al. Characterization of left ventricular diastolic function by tissue Doppler imaging and clinical status in children with hypertrophic cardiomyopathy. Circulation 2004;109:1756–1762.
[Article] [PubMed]29. Menon SC, Ackerman MJ, Cetta F, O'Leary PW, Eidem BW. Significance of left atrial volume in patients <20 years of age with hypertrophic cardiomyopathy. Am J Cardiol 2008;102:1390–1393.
[Article] [PubMed]30. Rickers C, Wilke NM, Jerosch-Herold M, Casey SA, Panse P, Panse N, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005;112:855–861.
[Article] [PubMed]31. Germans T, Wilde AA, Dijkmans PA, Chai W, Kamp O, Pinto YM, et al. Structural abnormalities of the inferoseptal left ventricular wall detected by cardiac magnetic resonance imaging in carriers of hypertrophic cardiomyopathy mutations. J Am Coll Cardiol 2006;48:2518–2523.
[Article] [PubMed]32. Price JF, Thomas AK, Grenier M, Eidem BW, O'Brian Smith E, Denfield SW, et al. B-type natriuretic peptide predicts adverse cardiovascular events in pediatric outpatients with chronic left ventricular systolic dysfunction. Circulation 2006;114:1063–1069.
[Article] [PubMed]33. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European pean Society of Cardiology. J Am Coll Cardiol 2007;50:1914–1931.
[Article] [PubMed]34. Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R, et al. Detection of viruses in myocardial tissues by polymerase chain reaction. evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol 2003;42:466–472.
[Article] [PubMed]35. Harmon WG, Sleeper LA, Cuniberti L, Messere J, Colan SD, Orav EJ, et al. Treating children with idiopathic dilated cardiomyopathy (from the Pediatric Cardiomyopathy Registry). Am J Cardiol 2009;104:281–286.
[Article] [PubMed] [PMC]36. Grenier MA, Fioravanti J, Truesdell SC, Mendelsohn AM, Vermilion RP, Lipshultz SE. Angiotensin-converting enzyme inhibitor therapy for ventricular dysfunction in infants, children and adolescents: a review. Prog Pediatr Cardiol 2000;12:91–111.
[Article] [PubMed]37. Ostman-Smith I. Hypertrophic cardiomyopathy in childhood and adolescence - strategies to prevent sudden death. Fundam Clin Pharmacol 2010;24:637–652.
[Article] [PubMed]38. Maskatia SA. Hypertrophic cardiomyopathy: infants, children, and adolescents. Congenit Heart Dis 2012;7:84–92.
[Article] [PubMed]39. Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997;336:775–785.
[Article] [PubMed]40. Frazier OH, Rose EA, Oz MC, Dembitsky W, McCarthy P, Radovancevic B, et al. Multicenter clinical evaluation of the HeartMate vented electric left ventricular assist system in patients awaiting heart transplantation. J Thorac Cardiovasc Surg 2001;122:1186–1195.
[Article] [PubMed]41. Liu J, Sluijter JP, Goumans MJ, Smits AM, van der Spoel T, Nathoe H, et al. Cell therapy for myocardial regeneration. Curr Mol Med 2009;9:287–298.
[Article] [PubMed]42. Vinge LE, Raake PW, Koch WJ. Gene therapy in heart failure. Circ Res 2008;102:1458–1470.
[Article] [PubMed] [PMC]43. Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H, et al. Improved clinical outcome after intracoronary administration of bone-marrow-derived progenitor cells in acute myocardial infarction: final 1-year results of the REPAIR- AMI trial. Eur Heart J 2006;27:2775–2783.
[Article] [PubMed]44. Decker JA, Rossano JW, Smith EO, Cannon B, Clunie SK, Gates C, et al. Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J Am Coll Cardiol 2009;54:250–254.
[Article] [PubMed]45. Ostman-Smith I, Wettrell G, Keeton B, Riesenfeld T, Holmgren D, Ergander U. Echocardiographic and electrocardiographic identification of those children with hypertrophic cardiomyopathy who should be considered at high-risk of dying suddenly. Cardiol Young 2005;15:632–642.
[Article] [PubMed]