About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 57(6); 2014
A chromosome 1q44 deletion in a 4-month-old girl; The first report in Korea
- Corresponding author: Young Youn Choi, MD. Department of Pediatrics, Chonnam National University Hospital, 42 Jebong-ro, Dong-gu, Gwangju 501-757, Korea. Tel: +82-62-220-6642, Fax: +82-62-222-6103, yychoi@chonnam.ac.kr
- Received May 07, 2013 Revised August 05, 2013 Accepted October 07, 2013
- Abstract
-
The deletion of the distal long arm of chromosome 1 is associated with a characteristic facial appearance and a pattern of associated malformations. Characteristic manifestations include a round face with prominent 'cupid's bow' and downturned corners of the mouth, thin vermilion borders of lips, a long upper lip with a smooth philtrum, a short and broad nose, epicanthal folds, apparently low-set ears, micrognathia, microcephaly, abnormal hands and feet, variable cardiac or genital anomalies, moderate to severe mental retardation, and growth retardation. Using fluorescent in situ hybridization (FISH) analysis to map precisely the deletion, we present a case of chromosome 1q44 deletion with craniofacial characteristics, multiple congenital anomalies, and growth and psychomotor retardation. In comparison with other reported cases of 1q43-44 deletion, the subject does not show hydrocephalus, seizure, syn- or polydactyly of hands, and a urogenital anomaly. However, an arachnoid cyst, pinpoint dimple on the midline of the forehead, a right-sided supernumerary nipple and auricular pit, polydactyly of the right foot, adducted thumb, and flexion restriction of the proximal interphalangeal joint with a simian line in both hands were observed additionally.
- Introduction
- Introduction
Deletion of the long arm of chromosome 1 has been reported very rarely since the first report of Mankinen et al.1). Specific phenotypic findings, resembling a distinct syndrome, are present in case with terminal deletion at chromosome 1q432). This deletion may result in a recognizable phenotype, including characteristic facial appearance, microcephaly, short neck, congenital malformations such as heart, skeletal and urogenital anomalies (in male patients), and severe psychomotor retardation with seizures1,2,3).Although there was one case of chromosome 1q deletion in Korea, the deletion site was more proximal (1q21.2)4). In this case, neonate with sialoblastoma and hepatoblastoma, confirmed by autopsy, was expired the day after birth. Here, we present the first case of chromosome 1q44 deletion in Korea, which was confirmed by fluorescent in situ hybridization (FISH) analysis, and compare it with other reported cases of 1q terminal deletion.
- Case report
- Case report
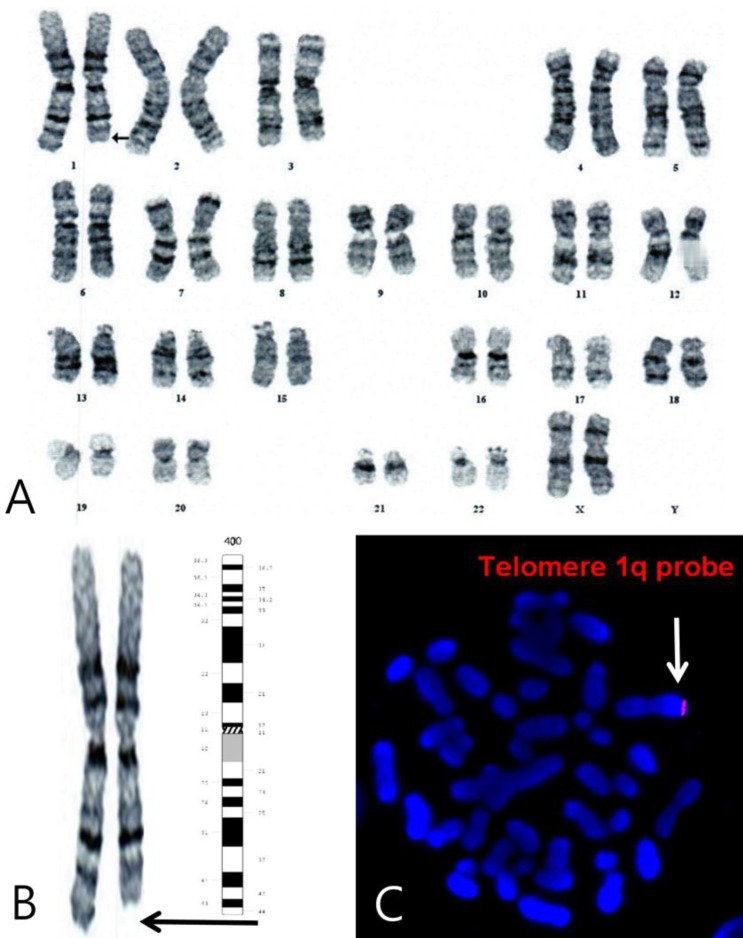
A 4-month-old girl was referred to Chonnam National University Hospital for the evaluation of microcephaly and polydactyly of her right foot. She was the first baby of a 28-year-old father and a 26-year-old mother. She was born at 38+6 weeks of gestation by cesarean section caused by a decreased amniotic fluid index (AFI) at a local hospital. Her birth weight was 2,800 g (25th-50th centile), length was 45.0 cm (3th-5th centile), and head circumference was 29.5 cm (<3rd centile). Apgar score was 9 at 1 minute and 10 at 5 minute. The baby's family history was negative for mental retardation or congenital anomaly. The mother's quadruple test was normal at the midtrimester. On the third trimester ultrasonography, small biparietal diameter and decreased amniotic fluid index (AFI=7) were noted.Her general appearance was dull and strange looking. Body weight was 5.9 kg (5th-10th centile), length was 56.8 cm (<3rd centile, 59.4 cm), and head circumference was 34.5 cm (<3rd centile, 38.4 cm). On physical examination, she showed microcephaly and prominent metopic suture with trigonocephaly, sparse and fine hair, round face, strabismus, upslanting palpebral fissure, periorbital fullness, hyperterolism with epicanthal folds, flat nasal bridge, short and broad nose, hypoplastic nares, smooth and long philtrum, thin vermilion borders, well formed 'cupid's bow' mouth with downturned corners, tucked in lower lip, micro/retrognathia, short webbed neck, and hypoplastic low-set ears with transverse groove on lobule with pit on limb of helix of the right ear. Additionally, a pinpoint dimple on the midline of the forehead (which was more prominent during her crying), small hands with simian line, flexion restriction of proximal interphalangeal (PIP) joint, tapered fingers and shortened, adducted thumbs, polydactyly of the right foot, and a supernumerary nipple on the right axillary region were also noted (Fig. 1A).There was a normal sinus rhythm with a grade of 2/6 systolic murmur on the left upper sternal border. Psychomotor retardation was noted. Muscle tone was decreased and could not lift her head nor make eye contact. Chest x-ray showed mild cardiomegaly and butterfly vertebra on the sixth thoracic spine (Fig. 2A). Her right foot showed postaxial polydactyly without a phalangeal bone. Echocardiographic findings showed secundum atrial septal defect (ASD) with moderate left-to-right shunt and right ventricle enlargement (Fig. 2B). Brain magnetic resonance imaging (MRI) revealed partial agenesis of the corpus callosum, delayed myelination, and a 1-cm cystic lesion in the left occipital convexity, suggesting arachnoid cyst (Fig. 3A, B). Because of her abnormal appearance, microcephaly, and developmental delay, a chromosome analysis was performed.The result of G-banding chromosomal analysis of peripheral blood lymphocytes was 46,XX,del(1)(q44) (Fig. 4A, B). Both parents and her younger brother's karyotypes were normal. FISH with probe 1q telomere (LSI single-color probe) was performed to verify the deletion site. The result showed that 100% of 200 interphase cells showed loss of a DiS3738 signal, e.g., 46, XX.ish del(1)(q44)(D1S3738-) (Fig. 4C).At 2 years and 2 months, her face was expressionless and had a dull appearance (Fig. 1B). Body weight was 8.8 kg (<3rd centile,10.2 kg), length was 72.1 cm (<3rd centile, 81.6 cm) and head circumference was 39.0 cm (<3rd centile, 45.4 cm). She had feeding difficulties due to gagging and vomiting. She could lift head, roll over, and sitting alone. She could creep but could not crawl. She was trying to grasp large object, transfer object from hand to hand. She distinguished strangers. The follow-up echocardiography showed closure of secundum ASD. And also, abdominal and renal ultrasonography revealed no abnormalities.
- Discussion
- Discussion
A reliable diagnosis of deletions of the distal part of chromosome 1q became available after the introduction of molecular cytogenetic methods. 1q44 is the most distal part of the long arm of chromosome 1: its genetic size is ~6 MB. Segment 1q43, which is approximately the same size, is more proximal part of 1q. There are numerous cytogenetic variants of the distal deletions 1q: interstitial deletions 1q43q44, terminal deletions 1q43qter, deletions involving only segment 1q44. The comparison of clinical findings between these groups did not show significant differences. The absence of the genetic material of 1q44 is the critical moment, and the additional absence of the genetic material of 1q43 does not seem to be clinically significant.Juberg et al.2) described a new case of 1q43 deletion and reviewed five other similar cases reported in the literature that initially delineated a new deletion syndrome. This delineation was confirmed and further extended by other authors3,5), who refined the distinctive terminal del (1)(q) syndrome. Among these, 22 patients were described as 1q42-qter deletion, 19 with 1q43-qter deletion, 6 with 1q44q-ter deletion, and 8 with interstitial deletion around 1q42-q443,5,6,7,8,9,10).Here, we present the first case of chromosome 1q44 deletion in Korea, confirmed by FISH analysis, and subsequently compare it with previously reported cases of terminal deletion of chromosome 1q (Table 1). All reported patients showed growth and psychomotor retardation. In addition, many patients showed CNS anomalies, such as agenesis/thin corpus callosum or hydrocephalus, hypotonia, seizure, autonomic dysfunction, and feeding difficulties. In our case, brain MRI revealed partial agenesis of corpus callosum, delayed myelination, and a 1-cm cystic lesion in the left occipital convexity, suggesting arachnoid cyst. However, seizure was not observed during the follow-up period.Many studies attempting to delineate the critical region for corpus callosum abnormalities (CCAs) have yielded inconsistent results. Boland et al.6) mapped the deletion and translocation breakpoints in 1q44, and suggested that haploinsufficiency of AKT3 is responsible for the CCAs. Otherwise, Nagamani et al.10) pointed out the AKT3 is important for normal brain development, and its haploinsufficiency causes microcephaly but not in CCAs. They implicated CEP170 and/or ZNF238 as novel genes causative for CCAs in patients with a terminal 1q deletion.On the other hands, Caliebe et al.11) suggested that HNRPU is a plausible candidate gene for the combination of developmental delay, hypotonia, CCAs, and seizures in patients with 1q44 deletions. The HNRPU gene encodes for heterogeneous nuclear ribonucleoprotein U, which is ubiquitously expressed. Ballif et al.12) concluded that HNRPU is the most plausible gene for the seizure phenotype, which apparently exhibits incomplete penetrance.Recently, Thierry et al.13) studied 11 unrelated patients with 1q44 microdeletion. All patients presented with moderate to severe intellectual disability, seizures and nonspecific craniofacial anomalies. By oligoarray-based comparative genomic hybridization (aCGH) covering the 1q44 region at a high resolution, they obtained a critical deleted region containing two coding genes-HNRNPU and FAM36A-and one noncoding gene-NCRNA00201. Nine of the 11 patients did not present with microcephaly or CCAs and carried a small deletion containing HNRNPU, FAM36A, and NCRNA00201 but not AKT3 and ZNF238, two centromeric genes. They suggest that HNRNPU, FAM36A, and NCRNA00201 are not major genes for microcephaly and CCAs but are good candidates for intellectual disability and seizures.Our case showed all of the characteristic craniofacial findings that were previously shown. In general, 1q terminal deletion syndromes accompany various congenital malformations, especially in the cardiac, urogenital and skeletal systems. In this case, ASD was existent while urogenital anomaly was not. Polydactyly was noted on the foot instead of the hand, and the butterfly vertebra on the sixth thoracic spine was shown. By coincidence, transverse groove with pit of the ear, a supernumerary nipple, and polydactyly of the foot were all noted on the right side.It is difficult to predict the long-term outlook for children with 1q deletions, since in many cases, this chromosome disorder has only fairly recently been diagnosed with certainty even among very few adults. Nonetheless, most children will need lifelong care and specialized medical support, and will only be able to achieve limited independence in carrying out their daily life. Many research reports describing 1q deletions focus on children with very large deletions, most of whom generally have a harder time. However, a larger number of children with deletions nearer the end of the chromosome at bands 1q43 and 1q44 appear to have better outlooks14).Genetic prognosis depends on the type of rearrangement. A significant part of terminal deletions may be a result of parental translocations: examination of parental chromosomes will be a prerequisite for further decisions. If one of the parents has a translocation, the recurrent risk will generally be relatively high. However, the risk for further children of the couple will be very low if parental karyotypes are normal15). Our case is a terminal de novo 1q44 deletion because both of her parents and her younger brother's karyotypes are normal.The limitation of our case, aCGH to obtain a critical deleted region containing various genes was not performed.In conclusion, the clinical phenotype of our patient has clear similarities with previous cases, suggesting a specific phenotype of patient with subtelomeric 1q terminal deletion. In comparing our case with other reported cases of 1q terminal deletion, hydrocephalus, seizure, syn- or polydactyly of hands, and urogenital anomaly are not shown. However, an arachnoid cyst, pinpoint dimple on the midline of the forehead, the right sided accessory nipple and auricular pit, polydactyly of the right foot, adducted thumb, and flexion restriction of PIP joints with simian line of both hands were noted additionally.
- Conflicts of interest
No potential conflict of interest relevant to this article was reported.
- References
- 1. Mankinen CB, Sears JW, Alvarez VR. Terminal (1)(q43) long-arm deletion of chromosome no. 1 in a three-year-old female. Birth Defects Orig Artic Ser 1976;12:131–136.2. Juberg RC, Haney NR, Stallard R. New deletion syndrome: 1q43. Am J Hum Genet 1981;33:455–463.
[PubMed] [PMC]3. van Bever Y, Rooms L, Laridon A, Reyniers E, van Luijk R, Scheers S, et al. Clinical report of a pure subtelomeric 1qter deletion in a boy with mental retardation and multiple anomalies adds further evidence for a specific phenotype. Am J Med Genet A 2005;135:91–95.
[PubMed]4. Huh CY, Choi HJ, Kim SB, Lee S, Lim SJ, Yang MH. 1 Case of chromosome 1q deletion with sialoblastoma and hepatoblastoma in neonate. Korean J Obstet Gynecol 1999;42:175–178.5. Gentile M, Di Carlo A, Volpe P, Pansini A, Nanna P, Valenzano MC, et al. FISH and cytogenetic characterization of a terminal chromosome 1q deletion: clinical case report and phenotypic implications. Am J Med Genet A 2003;117A:251–254.
[Article] [PubMed]6. Boland E, Clayton-Smith J, Woo VG, McKee S, Manson FD, Medne L, et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet 2007;81:292–303.
[Article] [PubMed] [PMC]7. Hill AD, Chang BS, Hill RS, Garraway LA, Bodell A, Sellers WR, et al. A 2-Mb critical region implicated in the microcephaly associated with terminal 1q deletion syndrome. Am J Med Genet A 2007;143A:1692–1698.
[Article] [PubMed]8. Merritt JL 2nd, Zou Y, Jalal SM, Michels VV. Delineation of the cryptic 1qter deletion phenotype. Am J Med Genet A 2007;143:599–603.9. Hiraki Y, Okamoto N, Ida T, Nakata Y, Kamada M, Kanemura Y, et al. Two new cases of pure 1q terminal deletion presenting with brain malformations. Am J Med Genet A 2008;146A:1241–1247.
[Article] [PubMed]10. Nagamani SC, Erez A, Bay C, Pettigrew A, Lalani SR, Herman K, et al. Delineation of a deletion region critical for corpus callosal abnormalities in chromosome 1q43-q44. Eur J Hum Genet 2012;20:176–179.
[Article] [PubMed]11. Caliebe A, Kroes HY, van der Smagt JJ, Martin-Subero JI, Tonnies H, van't Slot R, et al. Four patients with speech delay, seizures and variable corpus callosum thickness sharing a 0.440 Mb deletion in region 1q44 containing the HNRPU gene. Eur J Med Genet 2010;53:179–185.
[Article] [PubMed]12. Ballif BC, Rosenfeld JA, Traylor R, Theisen A, Bader PI, Ladda RL, et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet 2012;131:145–156.
[Article] [PubMed]13. Thierry G, Beneteau C, Pichon O, Flori E, Isidor B, Popelard F, et al. Molecular characterization of 1q44 microdeletion in 11 patients reveals three candidate genes for intellectual disability and seizures. Am J Med Genet A 2012;158A:1633–1640.
[Article] [PubMed]14. Understanding chromosome disorders. Unique. 1q4 deletions: from 1q42 and beyond [Internet]. Oxford: Unique/The Rare Chromosome Disorder Support Group, c1996-2014;cited 2013 Mar 10. Available from: http://www.rarechromo.org/information/chromosome%20%201/1q4%20deletions%20ftnw.pdf.15. Ioan DM, Maximilian C, Kleczkowska A, Fryns JP. Distal deletion of the long arm of chromosome number 1 (q43-->qter) associated with severe mental retardation and a nonspecific dysmorphic syndrome. Ann Genet 1992;35:167–169.
[PubMed]
Fig. 1
The characteristic phenotype of the chromosome 1q44 deletion. (A) At 4 months of age microcephaly, sparse and fine hair, a round face, pinpoint dimple on the midline of the forehead, flat nasal bridge, short and broad nose, hypoplastic nares, smooth and long philtrum, thin vermilion borders, a well formed 'cupid's bow' mouth with downturned corners, and short webbed neck were noted. Hypoplastic low-set ears with a transverse groove on the lobule and a pinpoint pit on the limb of the helix of the right ear, a supernumerary nipple on the right axillary side, polydactyly of the right foot, and simian creases on both hands were additionally noted. (B) At 2 years and 2 months of age, the patient's face was expressionless and had a dull appearance with severe growth and psychomotor retardation.
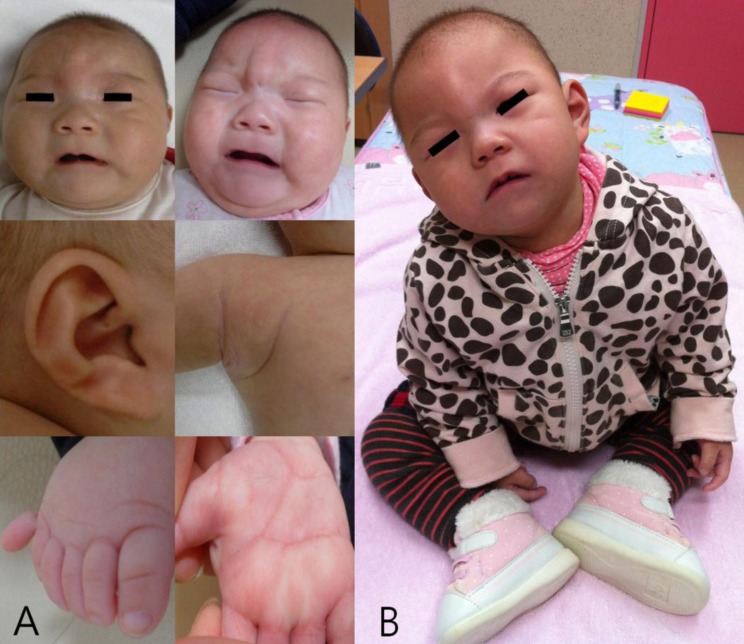
Fig. 2
Chest radiograph showed mild cardiomegaly and butterfly vertebra on the sixth thoracic segment of the spine (A, arrow), and echocardiography showed a secundum atrial septal defect (B, arrow).
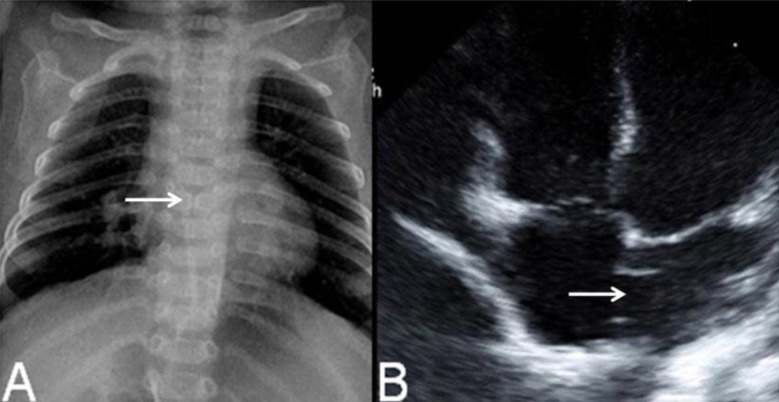
Fig. 3
Brain magnetic resonance imaging revealed partial agenesis of the corpus callosum (A, arrow) with arachnoid cyst (B, arrow).