About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 57(3); 2014
Lowe syndrome: a single center's experience in Korea
- Corresponding author: Han-Wook Yoo, MD, PhD. Division of Pediatric Endocrinology and Metabolism, Department of Pediatrics, Asan Medical Center Children's Hospital, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 138-736, Korea. Tel: +82-2-3010-3388, Fax: +82-2-473-3725, hwyoo@amc.seoul.kr
- Received September 01, 2013 Revised October 13, 2013 Accepted October 16, 2013
- Abstract
-
- Purpose
- Purpose
- Lowe syndrome is a rare, X-linked recessive disorder caused by mutations in the OCRL gene. It involves multiple anatomic systems, particularly the eyes, central nervous system, and kidneys, and leads to profound growth failure and global developmental delay. This study evaluated the clinical and genetic characteristics of Korean patients with Lowe syndrome.
- Methods
- Methods
- The clinical findings and results of genetic studies were reviewed for 12 male patients diagnosed with Lowe syndrome at a single medical institution.
- Results
- Results
- The mean age of the patients at presentation was 2.2 months (range, 0-4 months), although the diagnosis was delayed by a mean of 2.8 years (range, 0-9.7 years). The mean follow-up period was 9.0 years (range, 0.6-16.7 years). Nine mutations in OCRL were identified in 11 patients (92%), with three novel mutations. The main presentation was congenital cataract in both eyes necessitating early cataract removal in the 11 patients with impaired visual acuity. Profound short stature and developmental delay were observed in all patients, and seizures occurred in 50% of the patients. All patients suffered from proximal renal tubular dysfunction, and one patient developed chronic renal failure. Other manifestations included pathologic fracture (50%), cutaneous cysts (42%), and cryptorchidism (42%). However, there was no bleeding tendency, and none of the patients died during the study period.
- Conclusion
- Conclusion
- This study describes the clinical and genetic characteristics of Korean patients with Lowe syndrome. The observations are helpful for understanding the natural courses of Lowe syndrome and for appropriate genetic counseling.
- Introduction
- Introduction
Lowe syndrome (OculoCerebroRenal syndrome of Lowe) is an X-linked, recessive disorder characterized by congenital cataracts, mental retardation, and proximal renal tubular dysfunction (Fanconi syndrome)1). It is a rare disease with an estimated prevalence of 1-2 in 1,000,000 patients2).The clinical manifestations vary among affected individuals and also according to their age. Bilateral cataracts are present at birth and are associated with glaucoma in approximately half of the affected males, often resulting in progressive visual loss1). Global hypotonia is also noted soon after birth, and many patients exhibit mental retardation, stereotypic behavior, and seizure disorder3). During infancy and midchildhood, affected males have a variable spectrum of renal tubular dysfunction which causes aminoaciduria, metabolic acidosis, phosphaturia, and hypercalciuria. Later in life, some patients develop chronic renal insufficiency and end-stage renal disease between the second and fourth decades4). Renal rickets and osteomalacia due to renal dysfunction may lead to fractures, bone pain, and limited range of motion. Other clinical manifestations include facial dysmorphisms (frontal bossing, deep-set eyes, and protruding ears), dental malformation, superficial cysts, and failure to thrive5,6,7).This syndrome is caused by mutations in the OCRL gene on Xq25-q26, which contains 24 exons and encodes an inositol polyphosphate-5-phosphatase at the trans Golgi network as well as early endosomes8,9). OCRL has an essential role in cellular processes such as intracellular signaling, protein trafficking, and polymerization of the cytoskeleton actin. Its defect causes the several cellular defects underlying Lowe syndrome. Since 1952, when this disease was first recognized by Lowe et al.10), clinical and genetic studies have been reported in various populations. However, there have been a few studies in Korean patients and these reports only described the clinical characteristics of a few patients or mutations of the OCRL gene. In this study, we describe the detailed clinical features as well as the genetic characteristics in Korean patients with Lowe syndrome.
- Materials and methods
- Materials and methods
- 1. Study patients
- 1. Study patients
A total of 12, unrelated, male patients with Lowe syndrome were included in this study and had been referred from the Department of Pediatrics, Medical Genetics Center, Asan Medical Center Children's Hospital, Seoul, Korea, from December 1991 to April 2013. The demographic data, clinical manifestations, and the results of multidisciplinary studies, i.e., ophthalmologic, otolaryngologic, orthopedic, and urologic, were reviewed. Informed consent allowing genetic studies was obtained from all of the parents or guardians of all of the study patients.The clinical diagnosis of Lowe syndrome was based on the presence of all three of the following: (1) congenital bilateral cataracts; (2) renal Fanconi syndrome; and (3) involvement of the central nervous system, such as hypotonia and mental retardation1). Standard deviation scores (SDS) for height, weight, and body mass index (BMI) were calculated based on the data presented in the 2007 Korean Children and Adolescents Growth Standard11).- 2. Mutation analysis of the OCRL gene
- 2. Mutation analysis of the OCRL gene
Mutation analysis was carried out using genomic DNA from peripheral blood using a QuickGene DNA whole blood kit (Fujifilm, Tokyo, Japan). Twenty-three exons, including coding sequences of the OCRL gene and their intronic flanking sequences, were amplified by polymerase chain reaction (PCR) with 20 sets of primers. After PCR amplification with Go Taq polymerase (Promega, Madison, WI, USA), DNA sequencing was performed using the same primers as those in PCR and a BigDye Terminator V3.1 Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, USA).
- Results
- Results
- 1. Clinical characteristics at diagnosis
- 1. Clinical characteristics at diagnosis
The first presenting sign of Lowe syndrome in all 12 patients was bilateral congenital cataracts. The mean age at presentation was 2.2±1.8 months (0 to four months of age). However, clinical diagnosis of Lowe syndrome was delayed until 51.3±61.9 months of age (four months to 18 years of age). The interval from presentation to diagnosis was 34.1±34.8 months (0 to 116 months). At diagnosis, delayed development was seen in six patients (50%). In addition, proteinuria, nystagmus, seizure or sensorineuronal hearing loss were seen in the remaining six patients (Table 1).- 2. Genetic analysis of the OCRL gene
- 2. Genetic analysis of the OCRL gene
A total of nine different OCRL mutations, including three novel mutations, were identified in 11 patients (91.7%), although we could not find a mutation in the remaining patient (subject 12) (Table 1). Point mutations were one missense (p.Arg483Gln in subjects 3 and 8) and three nonsense (p.Arg483* in subject 1, p.Arg793* in subjects 4 and 6, and p.Arg678* in subject 11). Two, splicesite mutations, c.1551+1G>A and c.889-11G>A, were detected in subjects 5 and 7, respectively. The frameshift mutations, p.Leu723Trpfs*4 and p.Arg596fs*10, are expected to produce a prematurely terminated OCRL protein. c.2408_*5164inv refers to gross inversion of exons 21-23, which is also predicted to produce premature terminated protein. Three, novel mutations were c.2167_2174delTGGATGAA (p.Leu723Trpfs*4), c.1787_1788insGAAC (p.Arg596fs*10), and c.2408_*5164inv. The mutations were identified in exons 15 (three patients), 21 (three patients), and 17/18/19 (one patient each) in the order of frequency.Maternal carrier status was evaluated in seven families and was positive in two mothers (subjects 5 and 9). A positive family history was noted in subject 2 whose elder brother showed bilateral congenital cataract and sensorineuronal hearing loss. The elder brother died of sepsis at 29 months of age.- 3. Clinical course
- 3. Clinical course
All 12 patients survived during the follow-up period of 9.0±5.6 years (0.6 to 16.7 years).- 4. Physical growth
- 4. Physical growth
All patients presented with profoundly short stature (Table 2). The mean height SDS were -3.8±3.1 (-11.3 to -0.1) at diagnosis and -4.1±3.1 (-4.8 to 1.4) at the last follow-up visit, respectively. The mean BMI SDS was -0.8±1.7 (-4.8 to 1.4) at the last evaluation.There was marked delay in height and BMI in all cases at diagnosis and/or at the last follow-up visit. Linear growth velocities were below normal and short stature became evident by the age of one year in all 12 patients. The mean final height of four patients was 147.4±14.9 cm (128.7 to 165.0 cm), and the onset of puberty was delayed in two patients (subjects 1 and 4).- 5. Ocular manifestations
- 5. Ocular manifestations
Cataract removal surgery was performed in 11 patients at 3.6±1.6 months of age (one to seven months of age), and an intraocular lens was implanted in six (6/11, 55%) of these patients (Table 3). Nystagmus (9/12, 75%) and glaucoma (5/10, 50%) were also seen. Despite therapy, visual impairment worsened in all patients, and corneal keloid was found in three patients (3/10, 30%).- 6. Neurological manifestations
- 6. Neurological manifestations
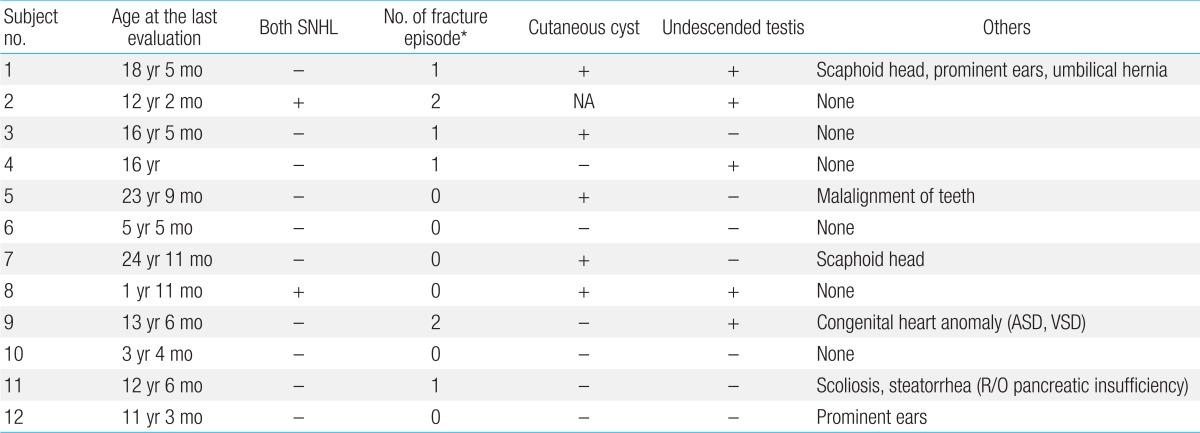
All patients experienced neonatal hypotonia with motor and cognitive developmental delay as well as mental retardation (Table 4). Six patients (50%) had clinical or electrical seizure disorders for which antiepileptic medication was needed. Brain magnetic resonance imaging was performed in seven patients. Patchy, white matter abnormalities were detected which were hyperintense on T2-weighted imaging (5/7, 71%), and those lesions were located in bilateral peritrigonal white matter in three patients (3/7, 43%; Fig. 1).- 7. Renal manifestations
- 7. Renal manifestations
Five patients whose urinary β2-microglobulin had been measured had albuminuria including low-molecular weight proteinuria (Table 5). In addition, proximal renal tubular acidosis or Fanconi syndrome was documented in all patients. Their estimated glomerular filtration rate (eGFR) was 82±24 mL/min/1.73m2 (47 to 144 mL/min/1.73m2), whereas one of these patients (subject 4) had renal insufficiency of eGFR 47 mL/min/1.73m2 at the age of 16 years12,13).Hypercalciuria (9/12, 75%), phosphaturia (6/12, 50%), renal rickets (9/12, 75%), hypokalemia (4/12, 33%), and nephrocalcinosis (7/12, 58%) were noted (Fig. 2). Sodium bicarbonate, citric acid, potassium, and phosphate replacement were required for metabolic and/or electrolyte disturbances, and calcitriol was given for renal rickets.- 8. Skeletal, integumental, and other manifestations
- 8. Skeletal, integumental, and other manifestations
Six patients (50%) experienced repeated pathologic bone fractures (Table 6). Pamidronate therapy was required for two patients (2/12, 17%) with severe osteoporosis seen on bone densitometry.Cutaneous cysts on the buttock or back were noted in five patients (5/12, 42%), and cryptorchidism was detected in five patients (5/12, 42%).An increased bleeding tendency was not observed during daily activities or at the time of surgery in any patient. Platelet counts, prothrombin time, and activated partial thromboplastin time were also normal in all cases.
- Discussion
- Discussion
Twelve patients in our study manifested the constellation of clinical features of Lowe syndrome. Bilateral congenital cataract is the earliest sign and requires surgery during infancy, although visual impairment can be reaggravated. Hypotonia, developmental delay, profoundly short stature, proximal renal tubular dysfunction or Fanconi syndrome and its complications such as renal rickets and nephrocalcinosis, were invariably seen in most of our study patients. However, only one patient had renal insufficiency. The small proportion of renal insufficiency in our patients is related to the short duration of the follow-up period as in Lowe syndrome chronic renal failure usually develops between the second and fourth decades of life14,15). However, its progression can occur even in early childhood, depending on tubular and glomerular dysfunction, as seen in nephropathic cystinosis. Recurrent pathologic bone fracture (50%), cutaneous cysts (42%), and cryptochidism (42%) were often observed in our patients. None of our patients had a bleeding tendency, in contrast to that seen in previously published reports16). All of our patients survived during the study period, although the survival rate remains to be re-estimated as the average life expectancy of patients with Lowe syndrome is approximately 40 years1).More than 200 different mutations in OCRL have been identified since the gene was discovered by Bailey Jr et al.17) in 1992. In our study, OCRL mutations were detected in 92% (11/12 alleles) of the patients, and which comparable to that seen in previous studies 2,4,16,18,19,20,21,22,23). Meanwhile, the most mutations, including nonsense, splice-site and frameshift mutations, are expected to produce prematurely truncated protein (10/11 alleles, 90.9%). This proportion of the truncating mutation is higher than that (63.6%) reported by Hichri et al.2). According to previous studies, the most frequent mutation site was exon 15 and the mutations were distributed in coding regions from exon 8 to exon 24, not in the first seven exons. In the present study, OCRL mutations were found in exon 15, 21, and 17/18/19, in order of frequency. The findings related to mutation sites are quite a contrast to those found in Dent-2 disease which was exclusively identified in the first seven exons of the OCRL gene and selectively shows renal proximal tubular dysfunction. It is known that the exons 1-7 of the coding region include the Pleckstrin Homology (PH) domain encoded by exons 2-5. Although the PH domain has a role as a major clathrin binding site, this suggests that the PH domain is not critical for the development of Lowe syndrome9).Meanwhile, three novel mutations were detected in our study. Two frameshift mutations, p.Leu723Trpfs*4 and p.Arg596fs*10, were identified in the ASH (ASPM, SPD-2, Hydin)/Rab binding domain spanning exons 17-19. The third novel mutation, c.2408_*5164inv, was gross inversion of exons 21-23 which mapped to a region in close interaction with the ASH domain. The ASH-RhoGAP (Rho GTPase activating) module regulates the majority of the protein-protein interactions for OCRL and has an important role in membrane recruitment9). Therefore, the frameshift or gross inversion mutations in this module that affect its interactions with binding partners, impair cellular localization and the stability of OCRL and are sufficient to cause disease.All patients with Lowe syndrome present in infancy with congenital cataract. However, the clinical diagnosis is established about two or three years later in most patients. Congenital bilateral cataract, neonatal hypotonia, and/or delayed development accompany other congenital syndromes due to chromosomal anomalies or prenatal infections1). In addition, the characteristic facial appearance and renal tubulopathy might not be obvious in the infantile or early childhood periods. Therefore, it is important to include Lowe syndrome in the differential diagnosis of a male infant with congenital cataract and hypotonia. When it is suspected, tubular proteinuria and elevation of aspartate transaminase, lactate dehydrogenase or creatine kinase are useful markers14). For confirmation of Lowe syndrome, the activities of the inositol polyphosphate-5-phosphatase can be measured in skin fibroblasts or genetic testing can be performed for the OCRL gene24). As for prenatal identification of Lowe syndrome, increased fetal nuchal translucency and the presence of bilateral lenticular opacities during the second trimester, as identified by detailed anomaly, are the important signs suggesting Lowe syndrome25,26). In these cases or in an informed family, prenatal testing can be performed by enzyme deficiency analysis and/or DNA analysis in fetal cells obtained by either chorionic villus sampling or by amniocentesis27). We could not obtain the information regarding prenatal ultrasonography in all of our patients. Meanwhile, in one case, the patient's mother planned another pregnancy and chorionic villus sampling was performed at 11 weeks and two days of gestation for antenatal exclusion of Lowe syndrome after the parent's prenatal consent (subject 11). As the mutational analysis showed that the fetus had a normal sequence of the OCRL gene, the parents decided to deliver the baby.Profound short stature and delayed puberty were both evident in our patients. Chronic renal failure and metabolic acidosis along with renal rickets are supposed to contribute to a patient's short stature and delayed puberty. Growth impairment is also a common feature in children with Fanconi syndrome28). In this respect, early identification and intervention for metabolic acidosis and renal rickets are important so as to deter growth impairment in children with Lowe syndrome. However, despite this aggressive therapy begun at an early age, growth retardation is inevitable in Lowe syndrome. However, there might be other elusive factors underlying the growth impairment.Although most of the clinical features of Korean Lowe syndrome patients did not differ from those reported in other countries, there are some differences in our study. According to Lasne et al.16), an abnormal rate of hemorrhagic events was found according to a retrospective clinical survey and prolonged closure times (CTs) were observed in the Platelet Function Analyzer-100 system, despite normal coagulation tests in six patients with Lowe syndrome. Our patients had the normal ranges of platelet counts, prothrombin time, and activated partial thromboplastin time, and no severe bleeding episode was experienced during their daily life activities or surgeries through the variable follow-up periods. As they may often need multiple surgeries, such as scoliosis reduction, hip surgery or eye surgery, bleeding diathesis should be thoroughly evaluated.Despite the extensive studies, it remains elusive how the OCRL gene mutations result in a variety of phenotypes of Lowe syndrome. As in our patients, most mutations are private. Sometimes clinical spectrum differs among patients with the same mutation (subjects 3 and 8 in Table 1). The underlying molecular mechanisms of these phenotypic heterogeneities are mainly elusive, although INPP5B, type II 5-phosphatase with an OCRL homolog with approximately 45% sequence homology and a similar structure, might have a role29). In the study of Bothwell et al.30), the OCRL knockout mice did not present phenotypes similar to those seen in Lowe syndrome, and the OCRL:INPP5B double knockout resulted in embryonic lethality, and leading to the hypothesis that INPP5B may compensate for OCRL in the mouse model. INPP5B shares a great portion of the interacting proteins with OCRL, except for clathrin and the endocytic clathrin adaptor AP-29). Differential expression of INPP5B in the relevant tissues may be related to the severity of the OCRL loss of the function phenotype. Therefore, further investigation regarding the compensatory role of INPP5B for OCRL in Lowe syndrome, will be needed and it will be helpful for developing potentially successful treatment of Lowe syndrome.Although we comprehensively reviewed the clinical and genetic findings of Korean patients with Lowe syndrome, our study has some limitations. First, the number of patients we reported in this study was too small to represent the general clinical and genetic features of Korean patients with Lowe syndrome and their genotype-phenotype correlations. In addition, the follow-up period was too short and variable among patients in order to assess the long-term natural course of Lowe syndrome in each patient. Because there is not a standardized, follow-up monitoring protocol, the clinical evaluation might have been incomplete in some patients.Until now, a few studies have been reported on Korean Lowe syndrome. Kim et al.31) provided a description of the clinical characteristics in two patients and other studies have focused on renal or integumental systems6,18). Meanwhile, our study first investigated the clinical features and genetic backgrounds of Korean patients with Lowe syndrome, as well as their heterogeneities. Long-term follow-up evaluations in a large number of patients will be needed in order to further investigate the clinical and genetic spectrum of Lowe syndrome in Korean patients.
- Acknowledgments
We thank the patients and their families for participating in this study which was supported by a grant from the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (Grant No. 2011-0019674).
- Conflicts of interest
No potential conflict of interest relevant to this article was reported.
- References
- 2. Hichri H, Rendu J, Monnier N, Coutton C, Dorseuil O, Poussou RV, et al. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat 2011;32:379–388.
[Article] [PubMed]3. Erdogan F, Ismailogullari S, Soyuer I, Ferahbas A, Poyrazoglu H. Different seizure types and skin lesions in oculocerebrorenal syndrome of Lowe. J Child Neurol 2007;22:427–431.
[Article] [PubMed]4. Bockenhauer D, Bokenkamp A, van't Hoff W, Levtchenko E, Kist-van Holthe JE, Tasic V, et al. Renal phenotype in Lowe syndrome: a selective proximal tubular dysfunction. Clin J Am Soc Nephrol 2008;3:1430–1436.
[Article] [PubMed] [PMC]5. Rodrigues Santos MT, Watanabe MM, Manzano FS, Lopes CH, Masiero D. Oculocerebrorenal Lowe syndrome: a literature review and two case reports. Spec Care Dentist 2007;27:108–111.
[Article] [PubMed]6. Won JH, Lee MJ, Park JS, Chung H, Kim JK, Shim JS. Multiple epidermal cysts in lowe syndrome. Ann Dermatol 2010;22:444–446.
[Article] [PubMed] [PMC]7. Maia ML, do Val ML, Genzani CP, Fernandes FA, de Andrade MC, Carvalhaes JT. Lowe syndrome: report of five cases. J Bras Nefrol 2010;32:216–222.
[Article] [PubMed]8. Silver DN, Lewis RA, Nussbaum RL. Mapping the Lowe oculocerebrorenal syndrome to Xq24-q26 by use of restriction fragment length polymorphisms. J Clin Invest 1987;79:282–285.
[Article] [PubMed] [PMC]9. Pirruccello M, De Camilli P. Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochem Sci 2012;37:134–143.
[Article] [PubMed] [PMC]10. Lowe CU, Terrey M, MacLachlan EA. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child 1952;83:164–184.
[PubMed]11. Korea Centers for Disease Control and Prevention, Division of Chronic Disease Surveillance, Committee for the Development of Growth Standard for Korean Children and Adolescents. Korean Pediatric Society, Committee for School Health and Public Health Statistics. 2007 Korean children and adolescents growth standard (commentary for the development of 2007 growth chart). Cheongwon: Korea Centers for Disease Control and Prevention, Division of Chronic Disease Surveillance, 2007.12. Schwartz GJ, Feld LG, Langford DJ. A simple estimate of glomerular filtration rate in full-term infants during the first year of life. J Pediatr 1984;104:849–854.
[Article] [PubMed]13. Schwartz GJ, Gauthier B. A simple estimate of glomerular filtration rate in adolescent boys. J Pediatr 1985;106:522–526.
[Article] [PubMed]14. Charnas LR, Bernardini I, Rader D, Hoeg JM, Gahl WA. Clinical and laboratory findings in the oculocerebrorenal syndrome of Lowe, with special reference to growth and renal function. N Engl J Med 1991;324:1318–1325.
[Article] [PubMed]15. Tricot L, Yahiaoui Y, Teixeira L, Benabdallah L, Rothschild E, Juquel JP, et al. End-stage renal failure in Lowe syndrome. Nephrol Dial Transplant 2003;18:1923–1925.
[Article] [PubMed]16. Lasne D, Baujat G, Mirault T, Lunardi J, Grelac F, Egot M, et al. Bleeding disorders in Lowe syndrome patients: evidence for a link between OCRL mutations and primary haemostasis disorders. Br J Haematol 2010;150:685–688.
[Article] [PubMed]17. Bailey LC Jr, Olivos IM, Leahey AM, Attree OF, Okabe I, Lewis RA, et al. Characterization of a candidate gene for OCRL [abstract]. Am J Hum Genet 1992;51(Suppl): A418. Cho HY, Lee BH, Choi HJ, Ha IS, Choi Y, Cheong HI. Renal manifestations of Dent disease and Lowe syndrome. Pediatr Nephrol 2008;23:243–249.
[Article] [PubMed]19. Addis M, Loi M, Lepiani C, Cau M, Melis MA. OCRL mutation analysis in Italian patients with Lowe syndrome. Hum Mutat 2004;23:524–525.
[Article]20. Satre V, Monnier N, Berthoin F, Ayuso C, Joannard A, Jouk PS, et al. Characterization of a germline mosaicism in families with Lowe syndrome, and identification of seven novel mutations in the OCRL1 gene. Am J Hum Genet 1999;65:68–76.
[Article] [PubMed] [PMC]21. Monnier N, Satre V, Lerouge E, Berthoin F, Lunardi J. OCRL1 mutation analysis in French Lowe syndrome patients: implications for molecular diagnosis strategy and genetic counseling. Hum Mutat 2000;16:157–165.
[Article] [PubMed]22. Lin T, Orrison BM, Leahey AM, Suchy SF, Bernard DJ, Lewis RA, et al. Spectrum of mutations in the OCRL1 gene in the Lowe oculocerebrorenal syndrome. Am J Hum Genet 1997;60:1384–1388.
[Article] [PubMed] [PMC]23. Kawano T, Indo Y, Nakazato H, Shimadzu M, Matsuda I. Oculocerebrorenal syndrome of Lowe: three mutations in the OCRL1 gene derived from three patients with different phenotypes. Am J Med Genet 1998;77:348–355.
[Article] [PubMed]24. Lowe M. Structure and function of the Lowe syndrome protein OCRL1. Traffic 2005;6:711–719.
[Article] [PubMed]25. Lin SY, Lee CN, Shih JC, Lin CH, Su YN. Two cases of Lowe syndrome presenting as increased fetal nuchal translucency. J Perinat Med 2011;39:483–485.
[PubMed]26. Daskalakis G, Anastasakis E, Lyberopoulos E, Antsaklis A. Prenatal detection of congenital cataract in a fetus with Lowe syndrome. J Obstet Gynaecol 2010;30:409–410.
[Article] [PubMed]27. Suchy SF, Lin T, Horwitz JA, O'Brien WE, Nussbaum RL. First report of prenatal biochemical diagnosis of Lowe syndrome. Prenat Diagn 1998;18:1117–1121.
[Article] [PubMed]28. Hsu SY, Tsai IJ, Tsau YK. Comparison of growth in primary Fanconi syndrome and proximal renal tubular acidosis. Pediatr Nephrol 2005;20:460–464.
[Article] [PubMed]29. Jefferson AB, Majerus PW. Properties of type II inositol polyphosphate 5-phosphatase. J Biol Chem 1995;270:9370–9377.
[Article] [PubMed]30. Bothwell SP, Chan E, Bernardini IM, Kuo YM, Gahl WA, Nussbaum RL. Mouse model for Lowe syndrome/Dent Disease 2 renal tubulopathy. J Am Soc Nephrol 2011;22:443–448.
[Article] [PubMed] [PMC]31. Kim SW, Yu YS, Kim IO, Cheong HI, Hwang YS, Choi Y. Two cases of oculocerebrorenal syndrome of Lowe. J Korean Pediatr Soc 1999;42:419–423.32. Chou YY, Chao SC, Chiou YY, Lin SJ. Identification of OCRL1 mutations in two Taiwanese Lowe syndrome patients. Acta Paediatr Taiwan 2005;46:226–229.
[PubMed]
Fig. 1
Axial brain magnetic resonance images of subject 12 at 4 years of age, showing white matter and the optic tract with high signal intensity on T2-weighted images.
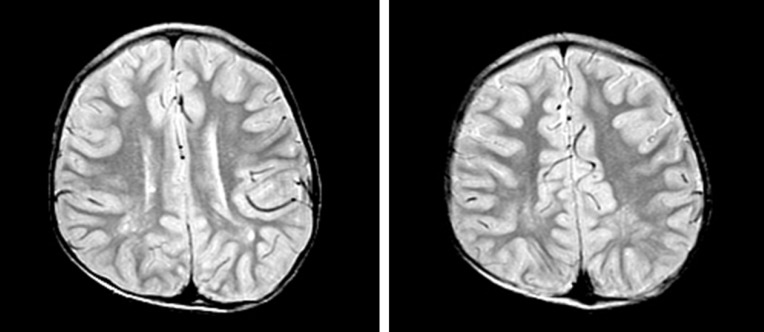
Fig. 2
Kidney ultrasonographic images of subject 2 at 10 years of age, showing echogenic medullae consistent with medullary nephrocarcinosis in both kidneys.
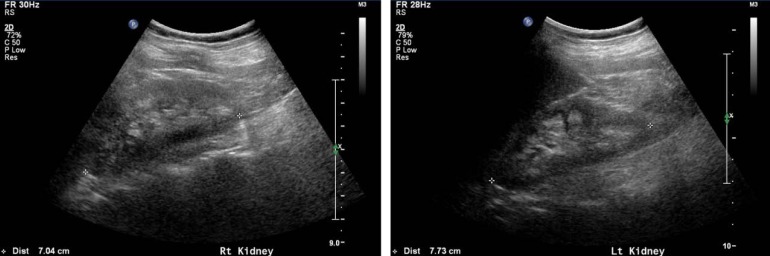
Table 1
Clinical findings and mutations of the OCRL gene at diagnosis in the patients with Lowe syndrome

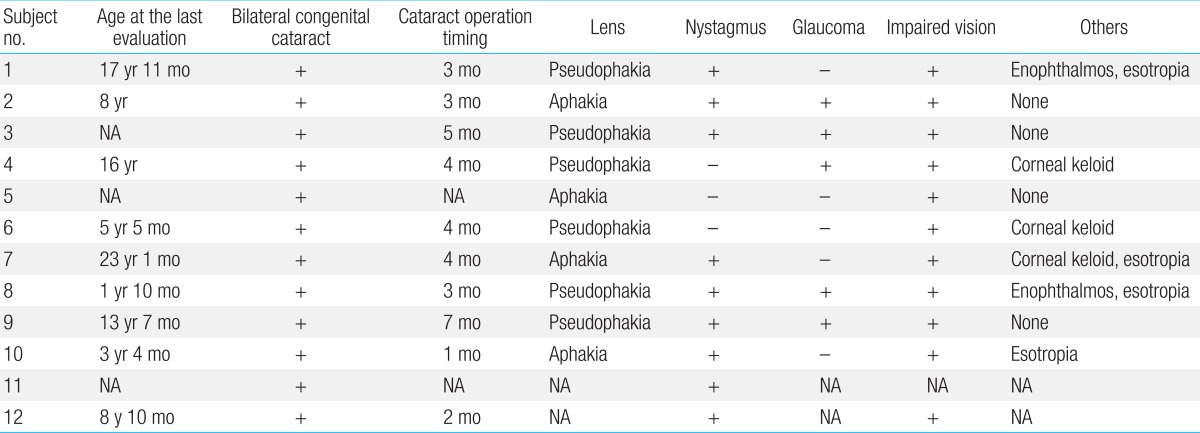
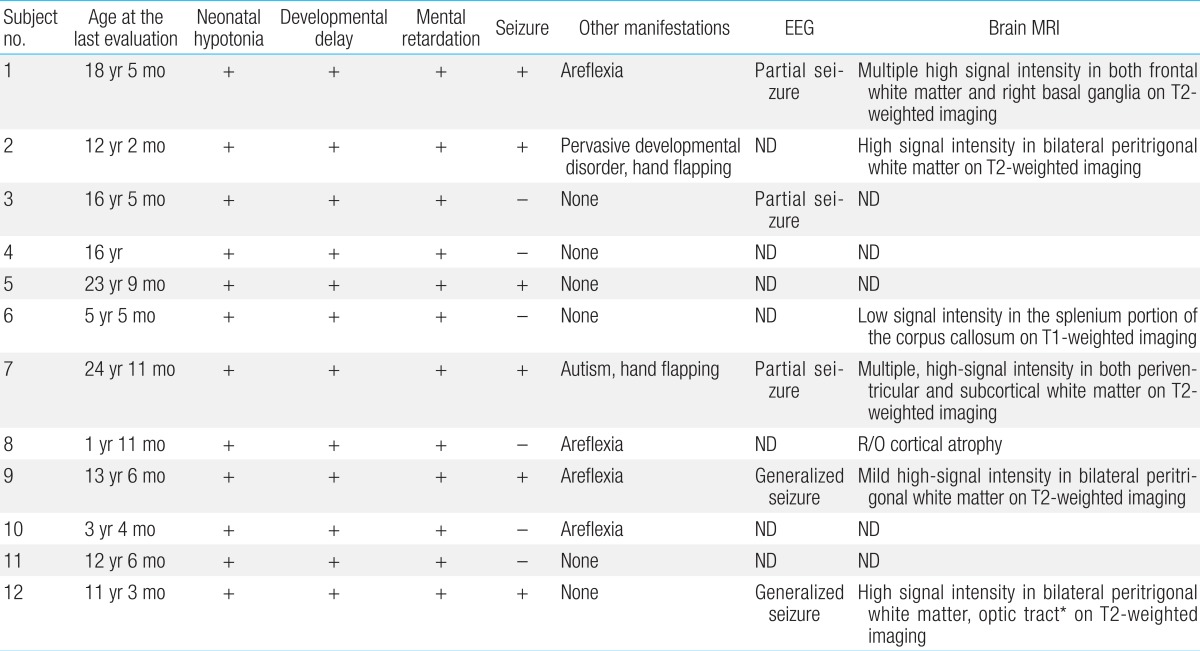
Table 5
Renal characteristics of the patients with Lowe syndrome
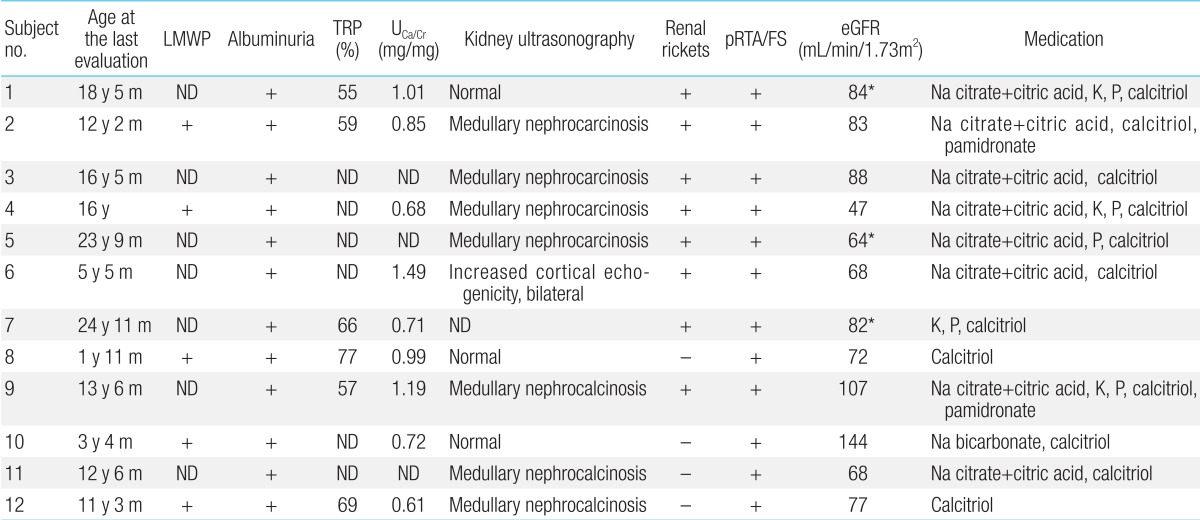
LMWP, low molecular weight proteinuria; TRP, renal tubular reabsorption of phosphate; UCa/Cr; urinary calcium/creatinine ratio; pRTA, proximal renal tubular acidosis; FS, Fanconi syndrome; eGFR, estimated glomerular filtration rate (using the Schwartz formula in patients under 18 years of age; and modification of diet in renal disease in patients [shown in *] over 18 years of age); ND, not done.