About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 59(1); 2016
Tuberculosis-associated hemophagocytic lymphohistiocytosis in adolescent diagnosed by polymerase chain reaction
- Corresponding author: Jung Sub Lim, MD, PhD. Department of Pediatrics, Korea Cancer Center Hospital, 75 Nowon-ro, Nowon-gu, Seoul 01812, Korea. Tel: +82-2-970-1224, Fax: +82-2-6008-5748, limjs5555@gmail.com
- Received November 27, 2013 Revised April 28, 2014 Accepted May 16, 2014
- Abstract
-
We present a case of tuberculosis-associated hemophagocytic lymphohistiocytosis in a 14-year-old girl. The patient presented with weight loss, malaise, fatigue, prolonged fever, and generalized lymphadenopathy. Laboratory investigation revealed pancytopenia (white blood cells, 2,020 cells/µL; hemoglobin, 10.2 g/dL; platelets, 52,000 cells/µL), hypertriglyceridemia (229 mg/dL), and hyperferritinemia (1,420 ng/mL). Bone marrow biopsy showed a hypocellular bone marrow with a large numbers of histiocytes and marked hemophagocytosis; based on these findings, she was diagnosed with hemophagocytic lymphohistiocytosis. Polymerase chain reaction (PCR) with both the bone marrow aspiration and sputum samples revealed the presence of Mycobacterium tuberculosis. Antitubercular therapy with immune modulation therapy including dexamethasone and intravenous immunoglobulin was initiated. The results of all laboratory tests including bone marrow biopsy and PCR with both the bone marrow aspiration and sputum samples were normalized after treatment. Thus, early bone marrow biopsy and the use of techniques such as PCR can avoid delays in diagnosis and improve the survival rates of patients with tuberculosis-associated hemophagocytic lymphohistiocytosis.
- Introduction
- Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an uncommon disorder characterized by a dysregulation of the activation and proliferation of macrophages, leading to uncontrolled phagocytosis of platelets, erythrocytes, lymphocytes, and their hematopoietic precursors throughout the reticuloendothelial system including in the bone marrow1,2).HLH was first described by Scott and Robb-Smith in 1939. They characterized by an acute onset and a progressively fatal course with fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, and widespread histiocytic infiltration. HLH was known to associate with various stimuli, such as infections, malignant neoplasms, and several other immune disorders. Of these, infection plays an important role in the etiology of the syndrome, and more than half of documented pediatric HLH cases occurred in countries in East Asia including Korea1,3).Mycobacterium tuberculosis is an important pathogen in Asia where the prevalence of tuberculosis is still high4), although Epstein-Barr virus (EBV) is known to be the most common cause of HLH among children. Over 50 cases of tuberculosis associated HLH have been published internationally5,6,7). Also several cases of tuberculosis associated HLH reported in adult, but not children in Korea8). The scarcity of reports on this condition may be due to the difficulties associated with diagnosing M. tuberculosis in HLH patients, who exhibit a high mortality rate. Here, we report the successful treatment of a female adolescent patient with HLH caused by tuberculosis, which was diagnosed early using polymerase chain reaction (PCR).
- Case report
- Case report
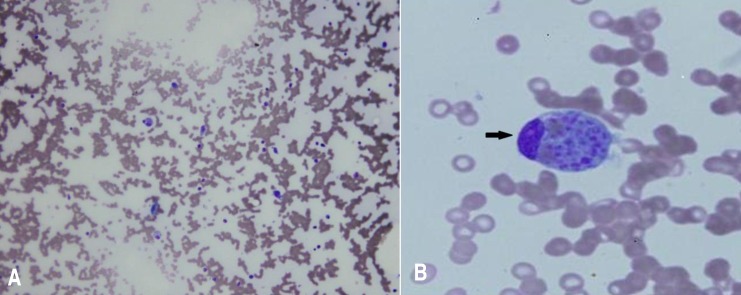
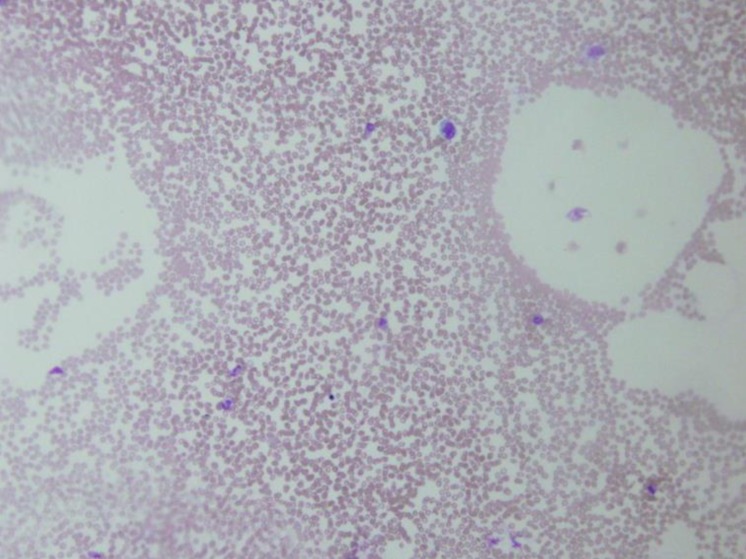
A 14-year-old girl was admitted to the hospital with fever, chills, malaise, fatigue, and a dull pain in the right subcostal area. She had lost 5 kg in bodyweight in the preceding 3 months. Ten days prior to presentation, she had complained of a remittent fever, with temperatures of up to 39.5℃. Two days prior to presentation, a dry cough and epistaxis had developed. There was no known history of contact with tuberculosis patients. Physical examination revealed a temperature of 38.8℃, cutaneous pallor, and mild tenderness on the right upper quadrant of the abdomen. Lymphadenopathy was detected on the both sides of the neck (a single matted lymph nodes in both side, more than 20 mm ×10 mm in the size that were firm with mild tenderness but not erytherma). There was no splenomegaly or hepatomegaly. Examination of the other systems revealed no abnormalities.Laboratory investigations undertaken at the initial assessment revealed microcytic hypochromic anemia (hemoglobin, 10.2 g/dL), leukocytopenia (2,020 cells/µL), thrombocytopenia (52,000 cells/µL) with poikilocytosis and atypical lymphocytosis identified in the peripheral smears. The erythrocyte sedimentation rate was increased to 26 mm/hr and the C-reactive protein level was slightly increased to 0.86 mg/L (normal range, <0.5 mg/L). Liver function tests revealed mild abnormalities (alanine transaminase, 54 U/L; aspartate aminotransferase, 153 U/L). We also observed mild hypocalcemia (8.0 mg/dL) with normal phosphorous (2.9 mg/dL) and alkaline phosphatase levels (61.0 U/L). Serum lactate dehydrogenase was 1535 U/L (reference range, 240–480 U/L). The chest radiograph did not reveal any suspicious abnormalities usually associated with pulmonary tuberculosis or pneumonia.During observation, the fever persisted with a remittent pattern. Pancytopenia was aggravated, and neutropenia was particularly aggravated (neutrophil count, 520 cells/µL). We evaluated the potential causes of the fever of unknown origin (FUO). We started treatment with cefepime for neutropenic fever on the second day. Further laboratory tests revealed elevated levels of lactate dehydrogenase (1,690 IU/L), hypertriglyceridemia (229 mg/dL), and hyperferritinemia (1,420 ng/mL). Multiple lymph node enlargements were detected on abdominal computed tomography (CT). We noted a hypocellular bone marrow with high numbers of histiocytes and marked hemophagocytosis on the bone marrow biopsy performed on day 7 after hospitalization (Fig. 1). The ratio of CD3CD4/CD3CD8 was reduced to 0.53 (normal range, 0.9–3.6) while CD16+CD56+ (natural killer cell activity) was within the normal range. However, caseous granuloma was not noted in the bone marrow specimen. The Mantoux test, acid-fast bacterium stain and culture of sputum revealed negative at that time. Results of several virus tests, including hepatitis A virus (negative for IgM and IgG), cytomegalovirus (negative for IgM, positive for IgG), EBV (negative for viral capsid antigen [VCA] IgM, early antigen R&D IgM and IgG, positive for VCA IgG and Epstein-Barr nuclear antigen IgG), and human immunodeficiency virus tests (negative for antihuman immunodeficiency virus), did not suggest acute infection. Only the venereal disease research laboratory test result was positive. However, the Treponema pallidum hemagglutination assay and fluorescent treponemal antibody-absorption test results were both negative. Results of fluorescent antinuclear antibody test was positive (1:1,280) but those of the rheumatoid arthritis factor test and anti-Smith antibody assay were both negative.On the basis of these findings, a diagnosis of HLH was made and treatment with high-dose dexamethasone (0.3 mg/kg/day for 7 days and tapering thereafter) and intravenous immunoglobulin (0.5 g/kg/day for 2 days) began on day 8 after hospitalization. At that time, the patient's activated partial thromboplastin time began to prolong to 44.3 seconds (normal range, 29.0–42.0 seconds) and fibrin degradation product was positive. After immune modulation, the fever subsided on day 9 after hospitalization, pancytopenia improved and ferritin level decreased to within the normal range. The PCR tests (AdvanSure TB/NTM real-time PCR using M. tuberculosis complex specific IS6110 and ITS specific primer) with both the bone marrow aspiration and sputum samples were reported to be positive for tuberculosis on day 10 after hospitalization. An antitubercular therapy regimen including 4 drugs—isoniazid 300 mg/day, rifampicin 450 mg/day, ethambutol 500 mg/day, and pyrazinamide 100 mg/day—was immediately started.On day 17 after hospitalization, the results of all laboratory tests were normalized. After 3 weeks of follow-up, cellularity was restored to normal level and no hemophagocytosis was observed at bone marrow biopsy (Fig. 2). Results of PCR with both the bone marrow aspiration and sputum samples were also negative for tuberculosis. The previously detected multiple lymph node enlargements were decreased in size on the follow-up abdominal CT. After 6 months of antitubercular therapy, the patient was healthy with no signs of disease.
- Discussion
- Discussion
Here, we report a rare case of tuberculosis-associated HLH in a female adolescent diagnosed using PCR performed with both the bone marrow and sputum samples. The patient was successfully treated with immune modulation and antitubercular therapy.In this case, we suspected disseminated tuberculosis based on clinical findings. Tuberculosis is widespread in Asian countries, including Korea, and is one of the important causes of FUO4,5,9). The clinical features of disseminated tuberculosis vary widely and are not often detected early. As the clinical symptoms of tuberculosis, which include fever, anorexia, weight loss, and weakness, are nonspecific and do not readily suggest a precise diagnosis. Pulmonary symptoms may or may not be present. If tuberculosis involves the extrapulmonary sites, generalized adenopathy, hepatomegaly, and splenomegaly may present. Only 10%–20% of patients with disseminated tuberculosis have a history of contact with tuberculosis. Definitive physical signs of disseminated tuberculosis are also rare. Fever may be the only sign of disseminated tuberculosis. Thus, disseminated tuberculosis should be suspected as one of the most frequent cause of FUO, especially in children, the elderly, and any group with decreased cell-mediated immunity. Most of the tuberculosis-associated hemophagocytic syndrome (HPS) cases in the literature were diagnosed with tuberculosis simultaneously. Furthermore, disseminated tuberculosis may be diagnosed after a long delay following the initial manifestation of hemophagocytosis10).Our patient had a fever for over 17 days, as well as pancytopenia, hypofibrinogenemia, hemophagocytosis of the bone marrow, and hyperferritinemia; which fulfilled the diagnostic criteria for HLH according to the 2004 Histiocyte Society Protocol entitled hemophagocytic lymphohistiocytosis1). Usually, symptoms of HPS include fever, hepatosplenomegaly, lymphadenopathy, and pancytopenia due to widespread histiocytic infiltration of the reticuloendothelial system, including the bone marrow. Furthermore, HLH is known to have an acute onset and a progressively increasing mortality risk as the condition develops, with an overall mortality rate of 52%1,2). One review reported 53 cases of tuberculosis associated HLH and 28 patients (52.8%) survived7). In another report of patients with immune deficiency, only 1 of the 6 patients (16.7%) survived11). Hemophagocytic syndrome-associated mortality is usually due to underlying disease or pancytopenia-associated complications. Eliopoulos et al reported that cause of death in tuberculosis-associated HLH include immunological disturbance and disseminated tuberculosis12).However, confirming the presence of tuberculosis is difficult because mycobacterial loads in the initial stage are very low and require several weeks of incubation before the organism can be isolated and identified, in addition, the sample yield is low. Thus, the Centers for Disease Control and Prevention recommended nucleic acid amplification test that aid early diagnosis13). Recently, one study examined the diagnostic utility of PCR for samples of bone marrow aspirate from patients with diverse clinical symptoms14). Using PCR, tuberculosis was detected in 70% of patients while the culture test detected tuberculosis in only 3.3% of patients. Clinical improvement with antitubercular therapy was observed in 85% of the patients with positive PCR results. In another study of 2,296 patients with suspected tuberculosis, the sensitivity, specificity, and positive predictive values of PCR were 97.2%, 100%, and 100% for smear-positive specimens and 75.3 %, 97.0%, and 47.5% for smear-negative specimens15).One study reported the survival rate among 29 patients who received therapy was 65.5% (12 out of 20 patients received a combination of immunomodulatory and antitubercular therapy and 7 out of 9 patients received antitubercular therapy alone) whereas no one who received no treatment survived. Moreover, the most failure of treatment resulted from the initiation of therapy late in the course of the illness5). Therefore, early diagnosis and antitubercular therapy besides the immunomodulatory therapy are important.To our knowledge, this is the first case of tuberculosis-associated HLH diagnosed by PCR and successfully treated using antitubercular therapy in Korean children. Considering that tuberculosis-associated HLH showed high mortality rate without antitubercular therapy and early bacteriological diagnosis of tuberculosis is difficult, we recommend tuberculosis PCR test for the early differential diagnosis of HLH patients.
- Conflicts of interest
Conflicts of interest: No potential conflict of interest relevant to this article was reported.
- References
- 1. Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis 2007;7:814–822.
[Article] [PubMed] [PMC]2. Verbsky JW, Grossman WJ. Hemophagocytic lymphohistiocytosis: diagnosis, pathophysiology, treatment, and future perspectives. Ann Med 2006;38:20–31.
[Article] [PubMed]3. Fisman DN. Hemophagocytic syndromes and infection. Emerg Infect Dis 2000;6:601–608.
[Article] [PubMed] [PMC]4. Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, Raviglione MC, et al. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med 2003;163:1009–1021.
[Article] [PubMed]5. Brastianos PK, Swanson JW, Torbenson M, Sperati J, Karakousis PC. Tuberculosis-associated haemophagocytic syndrome. Lancet Infect Dis 2006;6:447–454.
[Article] [PubMed]6. Balasubramanian S, Kaarthigeyan K, Aparna V, Srinivas S. Tuberculosis associated hemophagocytic syndrome in infancy. Indian Pediatr 2008;45:593–595.
[PubMed]7. Shea YF, Chan JF, Kwok WC, Hwang YY, Chan TC, Ni MY, et al. Haemophagocytic lymphohistiocytosis: an uncommon clinical presentation of tuberculosis. Hong Kong Med J 2012;18:517–525.
[PubMed]8. Kim HI, Kim SW, Chang HH, Lee JM, Kim NS, Kwon KT, et al. Causes and risk factors of mortality in adult patients with hemophagocytic syndrome. Infect Chemother 2012;44:51–55.
[Article]9. Cunha BA, Krakakis J, McDermott BP. Fever of unknown origin (FUO) caused by miliary tuberculosis: diagnostic significance of morning temperature spikes. Heart Lung 2009;38:77–82.
[Article] [PubMed]10. Dilber E, Erduran E, Kalyoncu M, Aynaci FM, Okten A, Ahmetoglu A. Hemophagocytic syndrome as an initial presentation of miliary tuberculosis without pulmonary findings. Scand J Infect Dis 2002;34:689–692.
[Article] [PubMed]11. Baraldes MA, Domingo P, Gonzalez MJ, Aventin A, Coll P. Tuberculosis-associated hemophagocytic syndrome in patients with acquired immunodeficiency syndrome. Arch Intern Med 1998;158:194–195.
[Article]12. Eliopoulos G, Vaiopoulos G, Kittas C, Fessas P. Tuberculosis associated hemophagocytic syndrome complicated with severe bone marrow failure and disseminated intravascular coagulation. Nouv Rev Fr Hematol 1992;34:273–276.
[PubMed]13. Diagnostic Standards and Classification of Tuberculosis in Adults and Children. This official statement of the American Thoracic Society and the Centers for Disease Control and Prevention was adopted by the ATS Board of Directors, July 1999. This statement was endorsed by the Council of the Infectious Disease Society of America, September 1999. Am J Respir Crit Care Med 2000;161(4 Pt 1): 1376–1395.
[Article] [PubMed]14. Escobedo-Jaimes L, Cicero-Sabido R, Criales-Cortez JL, Ramirez E, Romero M, Rivero V, et al. Evaluation of the polymerase chain reaction in the diagnosis of miliary tuberculosis in bone marrow smear. Int J Tuberc Lung Dis 2003;7:580–586.
[PubMed]