About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 57(9); 2014
Neurofibromatosis type 1: a single center's experience in Korea
- Corresponding author: Chong Kun Cheon, MD, PhD. Department of Pediatrics, Pusan National University Children's Hospital, Pusan National University School of Medicine, 20 Geumo-ro, Mulgeum-eup, Yangsan 626-700, Korea. Tel: +82-55-360-3158, Fax: +82-55-360-2181, chongkun@pusan.ac.kr
- Received December 03, 2013 Revised April 02, 2014 Accepted June 03, 2014
- Abstract
-
- Purpose
- Purpose
- Neurofibromatosis 1 (NF1) is an autosomal dominant condition caused by an NF1 gene mutation. NF1 is also a multisystem disorder that primarily affects the skin and nervous system. The goal of this study was to delineate the phenotypic characterization and assess the NF1 mutational spectrum in patients with NF1.
- Methods
- Methods
- A total of 42 patients, 14 females and 28 males, were enrolled in this study. Clinical manifestations and results of the genetic study were retrospectively reviewed.
- Results
- Results
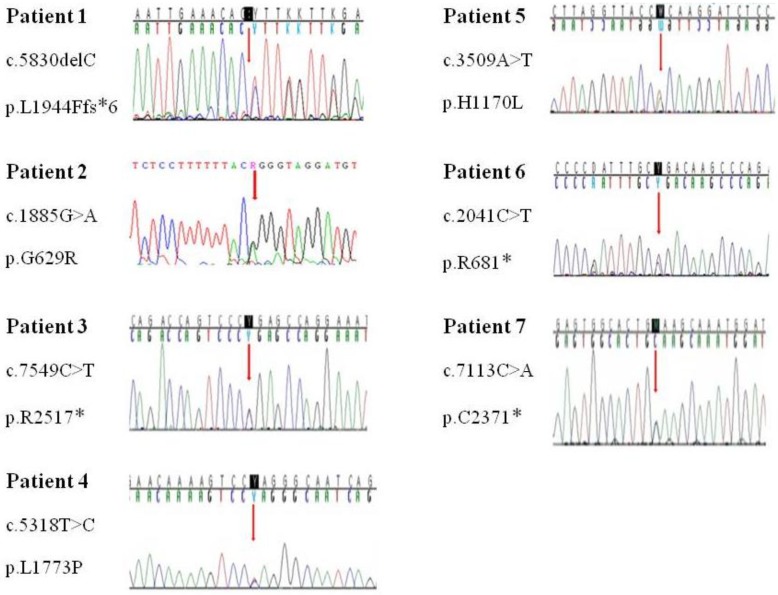
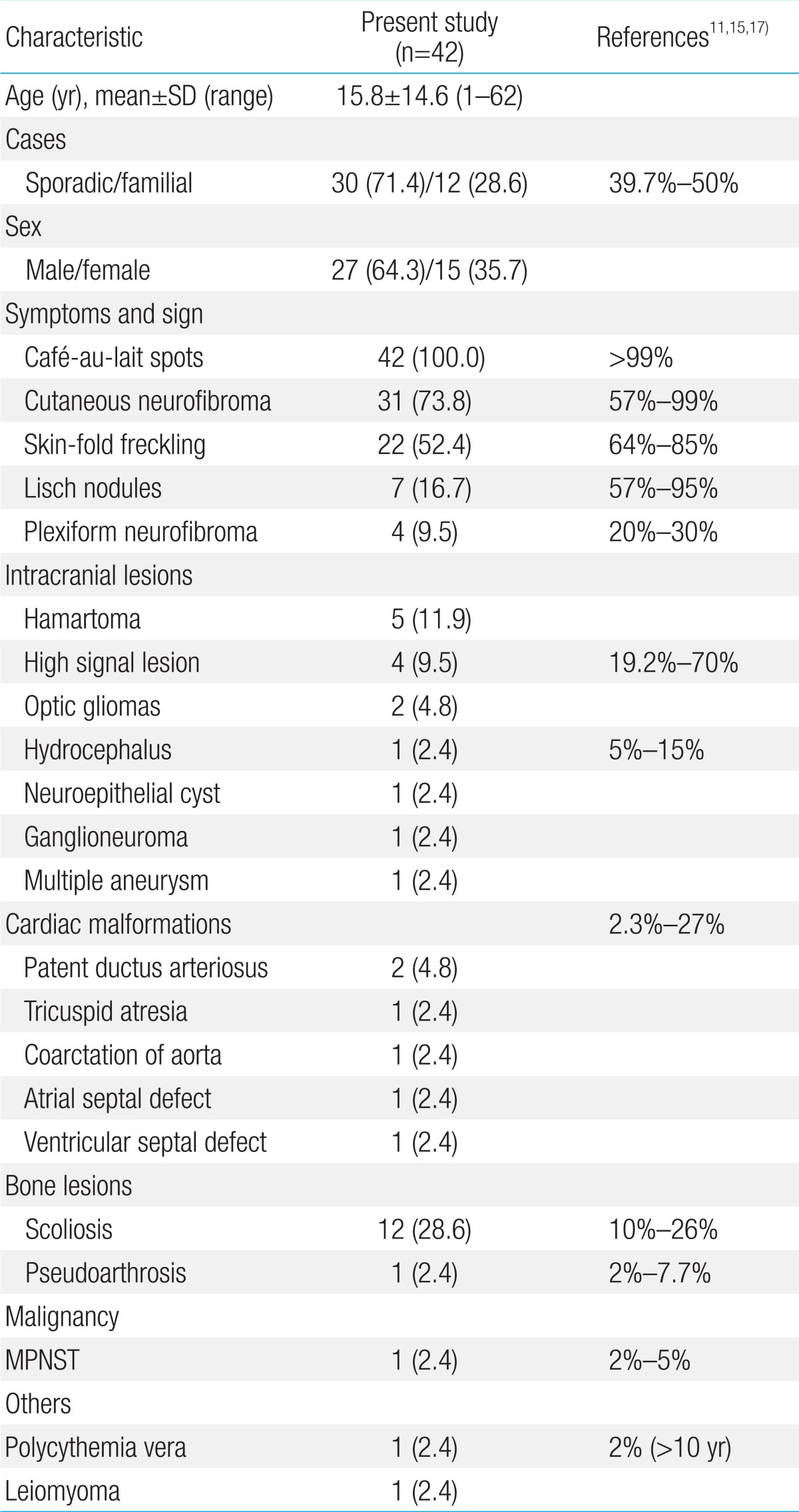
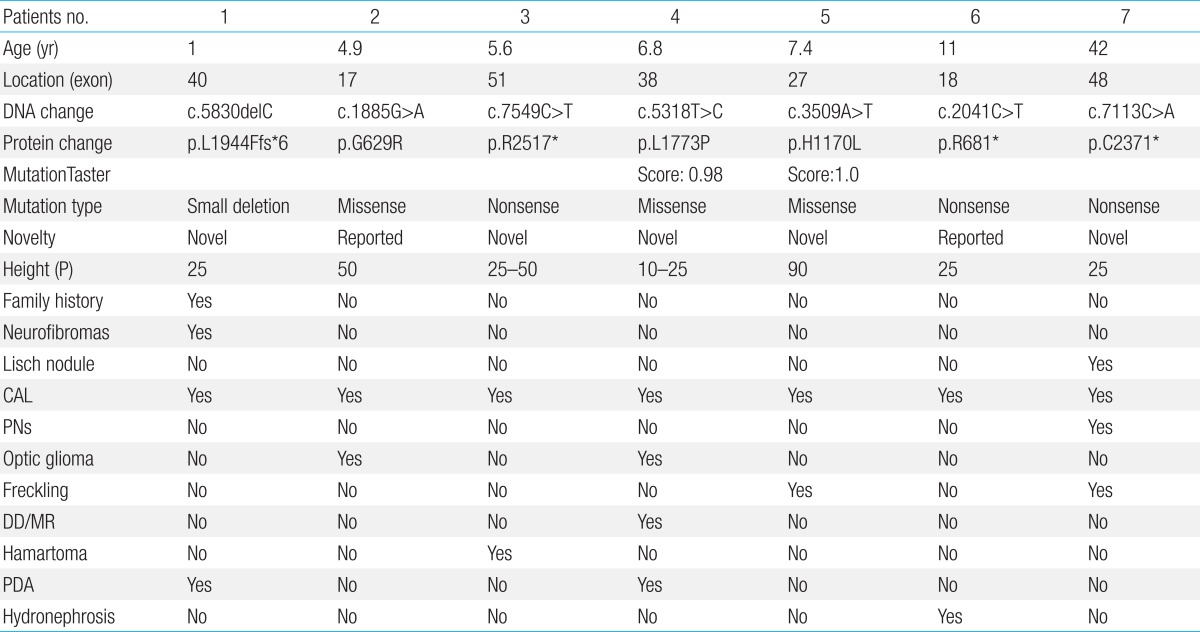
- Age of the patients at the time of NF1 diagnosis was 15.8±14.6 years (range, 1-62 years). Twelve patients (28.6%) had a family history of NF1. Among the 42 patients, Café-au-lait spots were shown in 42 (100%), neurofibroma in 31 (73.8%), freckling in 22 (52.4%), and Lisch nodules in seven (16.7%). The most common abnormal finding in the brain was hamartoma (20%). Mental retardation was observed in five patients (11.9%), seizures in one patient (2.4%), and plexiform neurofibromas (PNFs) in four patients (9.5%). One patient with PNFs died due to a malignant peripheral nerve sheath tumor in the chest cavity. Genetic analysis of seven patients identified six single base substitutions (three missense and three nonsense) and one small deletion. Among these mutations, five (71.4%) were novel (two missense mutations: p.Leu1773Pro, p.His1170Leu; two nonsense mutations: p.Arg2517*, p.Cys2371*; one small deletion: p.Leu1944Phefs*6).
- Conclusion
- Conclusion
- The clinical characteristics of 42 Korean patients with NF1 were extremely variable and the mutations of the NF1 gene were genetically heterogeneous with a high mutation-detection rate.
- Introduction
- Introduction
Neurofibromatosis type 1 (NF1) is a multisystem disorder primarily involving the skin and nervous system1). The manifestations of NF1 are extremely variable, even within a family2). Almost one half of all affected individuals have de novo mutations2). The criteria for diagnosis of NF1, developed by a National Institutes of Health (NIH) Consensus Conference in 1987, are established for routine clinical use3). The formal diagnosis of NF1 is made in an individual who has two or more of the following features in the absence of another diagnosis: Six or more Café-au-lait spots; freckling in the axillary or inguinal regions; two or more neurofibromas of any type or one plexiform neurofibroma (PNF); optic pathway gliomas; two or more Lisch nodules; distinctive bony lesions; or a first-degree family relative with NF1 as defined by the above criteria3). In addition, NF1 mutational analysis can clarify the diagnosis and will be helpful in describing the characteristics of molecular genetics of NF1 mutations4).In this study we sought to investigate various clinical manifestations and molecular genetic characteristics in Korean patients with NF1.
- Materials and methods
- Materials and methods
- 1. Study patients
- 1. Study patients
A total of 42 patients, 14 females and 28 males, were enrolled in this study from March, 2009 to September, 2012 at Pusan National University Children's Hospital. All NF1 patients met the diagnostic criteria proposed by the NIH and/or were genetically diagnosed with NF1. Blood samples were collected from the patients after consent was obtained from the parents. Mutation analysis and clinical review of these patients were approved by the Institutional Review Board at Pusan National University Yangsan Hospital. Clinical manifestations, clinical courses, and results of genetic study were retrospectively reviewed. The severe phenotype also includes one or of the following: malignant peripheral nerve sheath tumor (MPNST) and PNFs.- 2. Mutation analysis of the NF1 gene
- 2. Mutation analysis of the NF1 gene
Mutation analysis was carried out using genomic DNA from peripheral blood using a PUREGENE DNA isolation kit (Gentra, Minneapolis, MN, USA). All the coding exons and exon-intron boundaries of the NF1 gene were amplified by polymerase chain reaction (PCR) with 60 sets of primers. After PCR amplification with Taq polymerase (Promega, Madison, WI, USA), DNA sequencing was performed using the same primers as those in PCR and a BigDye Terminatore V3.0 Cycle Sequencing Ready reaction kit (Applied Biosystems, Foster City, CA, USA) according to manufacturer's instructions. The sequencing reaction mixtures were electrophoresed and analyzed using an ABI 3130xl Genetic analyzer (Applied Biosystems). The pathogenic probability for each sequence variation of novel mutations in the NF1 gene was predicted automatically by software MutationTaster (http://www.mutationtaster.org/).- 3. Statistical analysis
- 3. Statistical analysis
Statistical significance (P<0.05) was estimated by chi-square test for continuous variables and the Fisher exact test for nominal variables using SAS 9.2 (SAS Institute Inc., Cary, NC, USA).
- Results
- Results
- 1. Clinical manifestations of patients with NF1
- 1. Clinical manifestations of patients with NF1
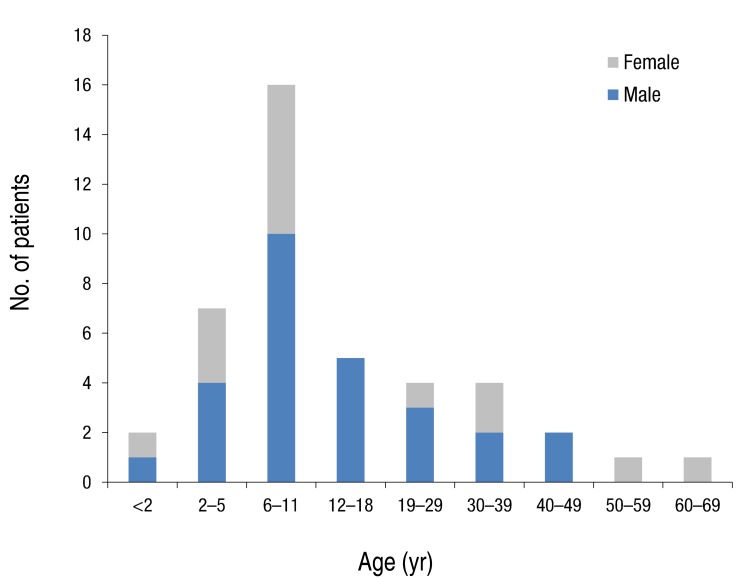
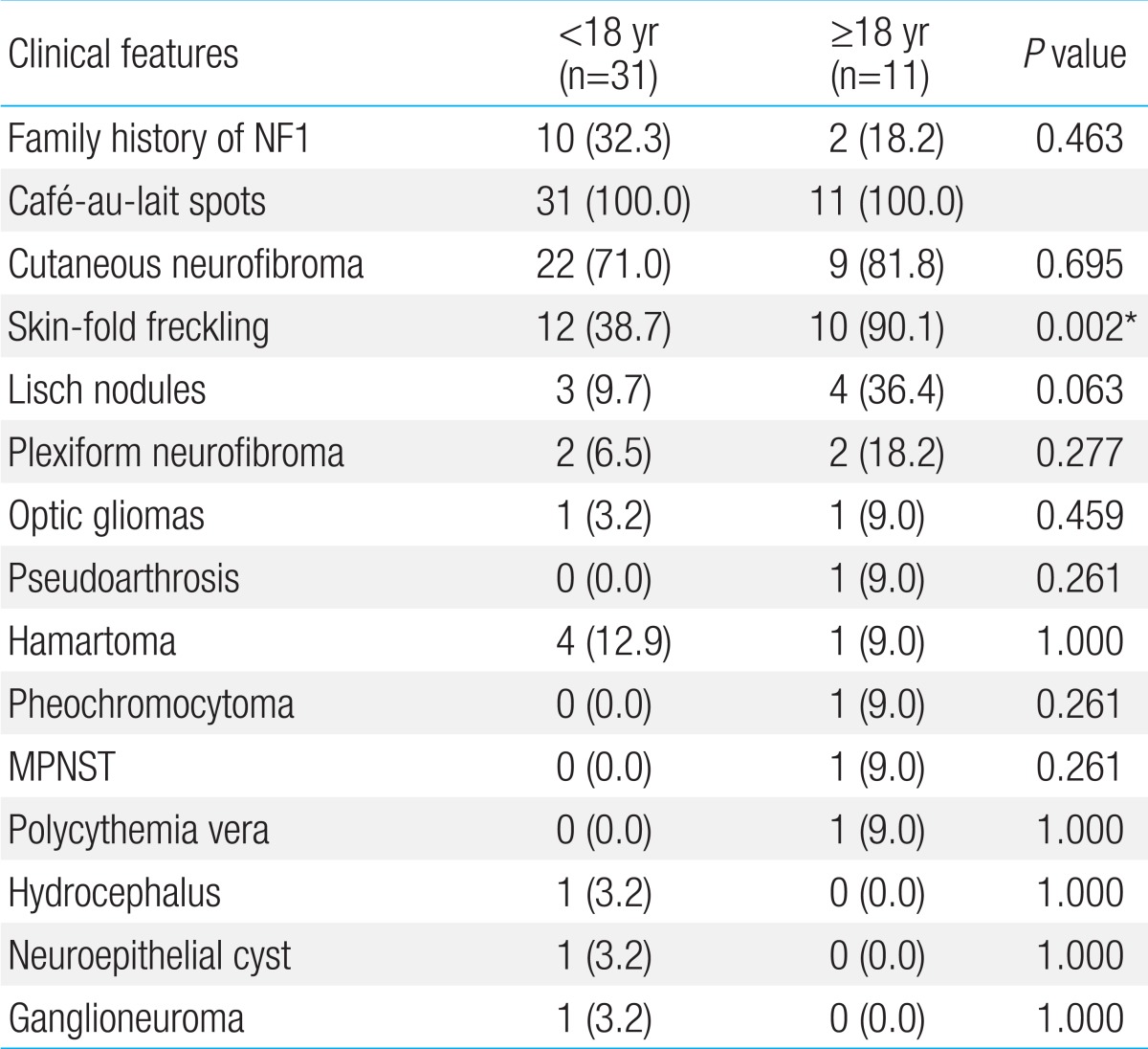
Forty two patients were diagnosed with NF1 during a period of three years six months. Detailed clinical delineation of all patients was performed and is summarized in Table 1. Twenty seven (64.3%) were male, and 15 (35.7%) were female. The age distribution of most patients, 16 (38%), was 6-11 years (Fig. 1). Age of the patients at the time of NF1 diagnosis was 15.8±14.6 years (range, 1-62 years). Twelve patients (28.6%) had family history of NF1. Among 42 patients, café-au-lait spots were shown in 42 (100%), neurofibroma in 31 (73.8%), and axillary freckling in 22 (52.4%), and Lisch nodules on the iris in seven (16.7%). A total of 25 patients with NF1 underwent brain magnetic resonance imaging (MRI); abnormal findings on MRI were detected in 13 patients (52%). The most common abnormal finding was hamartoma in five patients (20%). Of these, three patients had a cerebellum hamartoma, one patient had a cerebral hamartoma, and one patient had a basal ganglia hamartoma. Two patients (8%) had optic glioma, one patient (4%) had a neuroepithelial cyst on the right occipital area, and one patient (4%) had hydrocephalus. High signal lesions were observed in 4 patients (9.5%) and were particularly common in children. Multiple aneurysms were observed around the vertebral artery and Willis circle in one patient. Developmental delay or mental retardation was observed in five patients (11.9%), and seizure in one patient (2.4%). PNFs were observed in four patients (9.5%). The sites of PNFs included chest cavity, paraspinal area, left orbital cavity, and multiple neck, back, and nasal cavities. One of the patients with PNFs died due to MPNST in the chest cavity. Short stature was observed in three patients (7.1%), central precocious puberty in one patient (7.1%), and early puberty in one patient (7.1%). One 54-year-old adult presented with malignant hypertension due to pheochromocytoma and undergone right adrenalectomy. Spinal scoliosis was observed in 12 patients (28.6%), and pseudoarthrosis in one patient (2.4%). A cardiac anomaly was detected in three patients (7.1%), including patent ductus arteriosus (1), tricuspid valve atresia (1), and coarctation of the aorta and ventricular septal defect (1). Polycythemia vera accompanied to NF1 was observed in a 27-year-old male patient. Comparing two groups, patients under 18 and those over 18 years, skin-fold freckling was statistically more frequent in the group of patients over 18 years (P=0.002) (Table 2).- 2. Characteristics of molecular genetics of NF1
- 2. Characteristics of molecular genetics of NF1
Genetic analysis in seven patients revealed a nonsense mutation in three patients (42.9%), missense mutation in the three patients (42.9%), and deletion/frameshift mutation in one patient (14.2%) (Table 3). Among these mutations, 5 were novel ones (2 missense mutations: p.Leu1773Pro, p.His1170Leu; 2 nonsense mutation: p.Arg2517*, p.Cys2371*; 1 deletion/frameshift: p.Leu1944Phefs*6). The p.Gly629Arg and p.Arg681* mutations were previously reported mutations. No genetic mutations were detected in seven patients' parents. The NF1 mutations are evenly distributed over the entire exon (exon17-exon51). Each patient had a unique individual mutation, and no recurrent mutations were identified (Fig. 2). All the novel mutations in this study were tested in 100 normal alleles.
- Discussion
- Discussion
This study was performed to delineate phenotypic characterization and assess the NF1 mutational spectrum in patients with NF1. Recently, a number of correlations have been suggested, including: a mild of café-au-lait spots only in patients with an in-frame deletion; a more severe general phenotype in patients with nonsense mutations; patients with NF1 with a 1.4 Mb microduplication with mild learning difficulties, teeth and hair characteristics4,5). In this study, patient 4 with a novel missense mutation (p.L1773P) showed mild mental retardation, macrodontia and increased caries which could be shown in patients with NF1 with a 1.4 Mb microduplication. In addition, most of patients (2/3) with a nonsense mutation had a mild phenotype, except for one case with the severe phenotype. This result might suggest the complexity of the phenotype.Recent papers have reported that various clinical characteristics of NF1 might occur by more elements instead of a single gene, of which two modifier genes (GDNF, TLF) have been found6). Some features of NF1 may be present at birth and others are age-related manifestations. In this study, the incidence of Lisch nodules was low (16.7%). Lisch nodules increase in frequency with age and usually develop in early adolescence2). Young et al.7) reported that only 38% of children younger than 10 years had Lisch nodules, comparing with 77% of patients between 11 and 20 years and 94% of patients older than 21 years. The low frequency rate of Lisch nodules in this study may due to skewed distribution of age, 60% of patients younger than 11 years. In this study, there was no difference in the clinical features between children and adult patients, except for skin-fold freckling. Skin-fold freckling was more frequent in individuals over 18 years compared to individuals under 18 years (90.1% vs. 38.7%). Young children with NF1 may have only a few skinfold freckling in the axillary or inguinal regions and very young children may not show it, while older children and adults often have clusters of freckles and freckling in almost 90%8). This is the most observed in the axillary and inguinal regions, but can also be founded in submammary regions in women, in the back of the neck, or under the chin8). There might be several assumptions about low incidence rate of skin-fold freckling in patients under 18 years. The first, all patients under 5 years had no skin-fold freckling. So that skin-fold freckling occurred in twelve patients (60%) of ages between 6 and 18 years. The second, there might have statistically bias on cutoff of the age in dividing into two groups. The third, skin-fold freckling could be also observed in inguinal region and submammary regions. However, it is often overlooked in adolescent patients due to the reluctance of examination. To better understand of the difference of clinical presentation on according to age, it is important to have regular and periodic examination on patients.The NF1 gene, located on the long arm of chromosome 17 at band 11.2, spans over 60 exons distributed over more than 300 Kb9,10). The gene is considered to be a tumor suppressor gene and inactivation of neurofibromin leads to a disruption in cell growth regulation11). The NF1 gene shows high mutation rates regarding those observed in most other comparable inherited disease genes, and de novo mutations make up to almost 50% cases12). In this study, among seven patients tested with the NF1 gene, all patients had seven distinct mutations; two of these mutations were known mutations and five mutations were novel mutations. The putative functional relevance and pathogenicity of the two novel missense mutations (p.Leu1773Pro and p.His1170Leu) identified in the present study were predicted in silico using a MutationTaster software. The two novel missense mutations were predicted to affect shortened protein product or truncated protein product and to be disease-causing mutations and were not found in dbSNP (dbSNP137) and KPGP information (http://opengenome.net/). Jeong et al.13) reported that the mutation were evenly distributed across exon 3 through intron 47 of the NF1 gene with a novel mutation-detection rate (61.9%) in Korean patients. Ko et al.14) also reported that the mutations were equally distributed across exon 1 through intron 47 with a novel mutation-detection rate (30.8%), and no mutational hot spots were detected. This analysis revealed a wide spectrum of NF1 mutations in Korean patients and a genotype-phenotype analysis suggests that there is no clear relationship between specific mutations and clinical features. In this study, NF1 mutation are evenly distributed over the entire (exon 17-exon 51) with a novel mutation-detection rate (71.4%) and hot spot was not discovered.PNFs are associated with severity of disease and occur in 25% of patients with NF115). Prada et al.5) reported that pediatric patients with NF1 and PNFs had a higher mortality rate (3.2%) and suggested that nonsense mutations may predict a more severe general phenotype, which could include higher risk for PNFs. Among four patients with PNFs in this study, only one patient underwent genetic testing result in a novel nonsense mutation (p.Cys2371X). The patient had asymptomatic multiple nodular PNFs in the posterior neck, back, and right anterior nasal cavity and no combined complication or need for surgical intervention. However, Grobmyer et al.16) reported that PNFs can transform to MPNST with an estimated lifetime risk for patients with NF1 of approximately 10%. In the present study, malignancy-related death in the PNF group included hypovolemic shock caused by a massive hemothorax from a large thoracic MPNST during follow-up. Evans et al.17) reported that many patients with MPNST have nonresectable disease at diagnosis; Overall survival rate of affected individuals at 5 and 10 years was 20%-50% and 7.5%, respectively. Therefore, comprehensive evaluation, including genetic testing, in patients with NF1 might be helpful in prediction of high-risk group of tumor development.NF1 is associated with some malignancies including, rhabdomyosarcoma, leukemia, and other brain tumors17). In this study, all tumor were benign, apart from MPNST, of which hamartoma was the most common tumor (20%). However, Listernick et al.18) reported that the most common tumor in children with NF1 was the optic glioma, which was seen in 15% to 20% of patients. Although optic glioma in many patients with NF1 exhibit indolent behavior, approximately one-third to half of these tumors will cause clinical symptoms, including vision loss and precocious puberty18). In the present study, both of our patients presented with symptomatic optic glioma, including sudden onset visual loss and exophthalmos in whom two missense mutations, p.Leu1773Pro and p.Gly629Arg, were detected, respectively. This study has a limitation of the small number of patients who underwent genetic testing. Therefore, genotype-phenotype correlation in NF1 seems to be difficult to interpret. Additional molecular genetics study for large-scale national patients with NF1 and further functional and structural research of each NF1 mutation in specific regions are required.In summary, the clinical manifestations of 42 Korean patients with NF1 were extremely variable and the mutations of the NF1 gene were genetically heterogeneous with high a novel mutation-detection rate (71.4%). It is hoped that these data will contribute to a better understanding of the distinct molecular genetic characteristics of patients with NF1, and to expand the global pool of NF1 reports.
- Acknowledgments
We thank the patients and their families for participating in this study, which was supported by a 1-Year Research Grant of Pusan National University Yangsan Hospital.
- Conflicts of interest
No potential conflict of interest relevant to this article was reported.
- References
- 1. Hersh JH. American Academy of Pediatrics Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics 2008;121:633–642.
[Article] [PubMed]2. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med 2010;12:1–11.
[Article] [PubMed]3. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988;45:575–578.
[Article] [PubMed]4. Sharif S, Upadhyaya M, Ferner R, Majounie E, Shenton A, Baser M, et al. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J Med Genet 2011;48:256–260.
[Article] [PubMed]5. Prada CE, Rangwala FA, Martin LJ, Lovell AM, Saal HM, Schorry EK, et al. Pediatric plexiform neurofibromas: impact on morbidity and mortality in neurofibromatosis type 1. J Pediatr 2012;160:461–467.
[Article] [PubMed]6. Chong JA, Moran MM, Teichmann M, Kaczmarek JS, Roeder R, Clapham DE. TATA-binding protein (TBP)-like factor (TLF) is a functional regulator of transcription: reciprocal regulation of the neurofibromatosis type 1 and c-fos genes by TLF/TRF2 and TBP. Mol Cell Biol 2005;25:2632–2643.
[Article] [PubMed] [PMC]7. Young H, Hyman S, North K. Neurofibromatosis 1: clinical review and exceptions to the rules. J Child Neurol 2002;17:613–621.
[Article] [PubMed]8. DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000;105(3 Pt 1): 608–614.
[Article] [PubMed]9. Ledbetter DH, Rich DC, O'Connell P, Leppert M, Carey JC. Precise localization of NF1 to 17q11.2 by balanced translocation. Am J Hum Genet 1989;44:20–24.
[PubMed] [PMC]10. de Goede-Bolder A, Cnossen MH, Dooijes D, van den Ouweland AM, Niermeijer MF. From gene to disease; neurofibromatosis type 1. Ned Tijdschr Geneeskd 2001;145:1736–1738.
[PubMed]11. Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell 2001;104:593–604.
[Article] [PubMed]12. Valero MC, Pascual-Castroviejo I, Velasco E, Moreno F, Hernandez-Chico C. Identification of de novo deletions at the NF1 gene: no preferential paternal origin and phenotypic analysis of patients. Hum Genet 1997;99:720–726.
[Article] [PubMed]13. Jeong SY, Park SJ, Kim HJ. The spectrum of NF1 mutations in Korean patients with neurofibromatosis type 1. J Korean Med Sci 2006;21:107–112.
[Article] [PubMed] [PMC]14. Ko JM, Sohn YB, Jeong SY, Kim HJ, Messiaen LM. Mutation spectrum of NF1 and clinical characteristics in 78 Korean patients with neurofibromatosis type 1. Pediatr Neurol 2013;48:447–453.
[Article] [PubMed]15. Khan AK, Deb S, Ray DK, Mandal SN, Mukhopadhaya S, Mandal S. Diffuse neurofibroma of scalp. Neurol India 2002;50:516–517.
[PubMed]16. Grobmyer SR, Reith JD, Shahlaee A, Bush CH, Hochwald SN. Malignant peripheral nerve sheath tumor: molecular pathogenesis and current management considerations. J Surg Oncol 2008;97:340–349.
[Article] [PubMed]