 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 63(6); 2020
Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability
-
Ji Yoon Han, MD, PhD, In Goo Lee, MD, PhD

- Corresponding author: In Goo Lee, MD, PhD. Department of Pediatrics, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, 222, Banpodaero, Seocho-gu, Seoul 06591, Korea E-mail iglee@catholic.ac.kr
- Received July 15, 2019 Revised October 7, 2019 Accepted October 24, 2019
- Abstract
-
Developments in next-generation sequencing (NGS) techogies have assisted in clarifying the diagnosis and treatment of developmental delay/intellectual disability (DD/ID) via molecular genetic testing. Advances in DNA sequencing technology have not only allowed the evolution of targeted panels but also, and more currently enabled genome-wide analyses to progress from research era to clinical practice. Broad acceptance of accuracy- guided targeted gene panel, whole-exome sequencing (WES), and whole-genome sequencing (WGS) for DD/ID need prospective analyses of the increasing cost-effectiveness versus conventional genetic testing. Choosing the appropriate sequencing method requires individual planning. Data are required to guide best-practice recommendations for genomic testing, regarding various clinical phenotypes in an etiologic approach. Targeted panel testing may be recommended as a first-tier testing approach for children with DD/ID. Family-based trio testing by WES/WGS can be used as a second test for DD/ ID in undiagnosed children who previously tested negative on a targeted panel. The role of NGS in molecular diagnostics, treatment, prediction of prognosis will continue to increase further in the coming years. Given the rapid pace of changes in the past 10 years, all medical providers should be aware of the changes in the transformative genetics field.
- Introduction
- Introduction
Developmental delay (DD) and intellectual disability (ID) affect 1% to 3% of children and about have a genetic cause in about half of all cases [1-3]. The genetic etiologies include chromosomal abnormalities, copy number variants (CNVs), or mutations. Genetic testing aims to identify a particular disorder and educate the family about neurological problems, comorbidities, expected future requirements, and possible treatment options. First-generation DNA sequencing with chin-terminating inhibitors discovered by Sanger in 1977, established many genetic etiologies and has been generally used for over 30 years in research era and clinical settings [4]. In 2004, next-generation sequencing (NGS) has begun to replace Sanger sequencing since it can sequence a large number of genes at once, the whole exome (protein-coding regions) or entire genome, and advances in NGS have resulted in increased detection of the genetic etiology of DD/ID. In recent years, NGS technologies have been widely applied in pediatric neurology clinics. Currently, three NGS approaches are in use: targeted gene panels (TGPs), whole-exome sequencing (WES), and whole-genome sequencing (WGS), and all three approaches have superior abilities to sequence large amounts of DNA compared to Sanger sequencing [5].High-throughput DNA sequencing technology has rapidly expanded over the past 15 years, and more advanced methods are steadily being commercialized [6]. Advances in NGS technologies have enormously accelerated the pace of data generation in the order of hundreds of giga-bases of nucleotides sequenced per machine run and reduced the sequencing cost by over 5 orders of magnitude. NGS technologies are widely applied clinically to facilitate insight into the complexity of disease mechanisms and contribute to developments in early diagnostics, prediction of prognosis, and proper management of tailored medicine [7]. Therefore, sequencing by NGS technology has rapidly entered the clinical field and NGS technology has overcome many barriers in research of rare Mendelian diseases.
- Algorithmic approach for children with DD/ID
- Algorithmic approach for children with DD/ID
Diagnostic evaluation to identify an etiology in children with global DD/ID (GDD/ID) is guided by a careful 3-generation family history, using “head to toe” approach that includes neurologic examination and evaluation for dysmorphic features or other congenital anomalies), and availability of specific tests [8]. A systemic diagnostic approach is required for children with GDD/ID to determine a particular underlying genetic etiology [2]. A broad clinical assessment using stepwise evaluation process remains the key to planning survey among children with DD/ID [9]. Chromosomal microarray analysis (CMA) is considered the first-tier test for genetic diagnosis of undiagnosed DD/ID [10]. Estimates suggest that up to 50% of children are unsuccessful to accept molecular diagnosis after extensive testing and enter on a diagnostic odyssey which is both time-consuming and expensive for health-care providers [11]. The diagnostic rate of children with clinical presentations such as DD/ID and autism spectrum disorders ranged from 5% to 50% in a tertiary center [12-15]. The traditional approach to diagnosing children with DD/ID is on the threshold of a paradigm shift due to the utilization of NGS (Fig. 1), and the use of TGP, WES, and WGS by NGS is now widespread. Many researchers have reported using gene panels, WES, and WGS in children with DD/ID for whom the etiology was unknown [16]. The efficacy of NGS approaches for causative gene identification was first shown in undiagnosed rare syndromes [17-19]. However, considerable discussion is needed regarding its optimal application of NGS in the diagnosis of DD/ID in children.
- Targeted gene panels
- Targeted gene panels
TGP for specific disease categories is used to determine known disease-causative genes. The focus on a restricted set of genes enables greater depth of coverage which increases analytic sensitivity and specificity. Better depth of coverage improves the confidence of heterozygous calls and detection of mosaicism or low-level heterogeneity in mitochondrial or oncology applications. The cost of TGP varies, but it is generally lower than that of WES or WGS.TGP tests via NGS may be preferable choice for establishing a genetic diagnosis of DD/ID [20]. Studies have suggested TGP as the first-tier testing approach compared with WES for several diseases based on diagnostic rate, coverage, depth, and cost reduction [21-25]. TGP produces a diagnostic rate similar to that of WES, while recent reports showed the efficacy of NGS testing for specific neurologic diseases [26-28]. Targeted panel sequencing is not indicated for children with multiple features, overlapping conditions, or less differentiated clinical phenotypes [29].TGP can be customized to cover only specific genomic regions of significance, which simplifies the workflow and produce results within a few days [30]. The experimenter can handle thousands of samples on a single sequencing course. Clinical expertise in DD/ID and the relevant genetic knowledge are important in choosing the suitable genes for the panels. The target list of gene panels should be regularly edited based on recent findings reflecting newly established associations of additional genes for DD/ID [31].TGP has been preferred because of low sequencing cost, short turnaround time, and low rates of nonspecific or incidental results, whereas only about 10% of the mutations are perceptible by WES were included. Among all the three NGS approaches, TGP has the lowest false-negative call rate for a categorized set of disease-associated genes [32]. The disadvantage of TGP over WES and WGS is its limited use for discovering new genes. If the panel test does not identify causative genes, WES or WGS can be considered the most comprehensive second-tier test [23].
- Whole-exome sequencing
- Whole-exome sequencing
WES can be used concurrently interpret the protein-coding exons of thousands of genes via NGS techniques, with reads representing about 1%–2% of the genome. Sequencing a patient’s exome and comparing it with a normal reference sequence, variations in and the causes of an individual’s medical concern and the cause of the medical disorders can be found.WES is generally used in patients who have tested negative on previous targeted panels of complex phenotypes without an obvious differential diagnosis, and it can be a second-tier option in cases where a molecular diagnosis is not applicable. Recently, WES is increasingly applicate as the premier and first-line test for rare and undiagnosed Mendelian disorders [33-36]. The reported yield of genetic causes for DD/ID by WES was 16% to 45%.37- [39] Genome-wide analyses using clinical WES have greatly increased the diagnostic yield of patients with suspected genetic diseases [40,41]. When the initial WES does identify an etiology, reanalysis of the sequencing data can bring resolution in 10% to 30% of cases, which is related to the advances in NGS technological advances and new disease associations [42]. More than 97% of variants observed by Sanger sequencing can be identified via WES, and with increased information about of mosaicism, WES efficiently performs extensive molecular evaluations.However, WES does not provide a “whole” coverage, and its capture step of WES results in greater coverage depth variability. The limitations of WES as a diagnostic method relate to developing knowledge of variant interpretation. although its coverage and price are between those of TGP and WGS (Fig. 2). WES depth of coverage is inconsistent with diagnostic sensitivity, which may be lower than that of TGP, since a considerable number of exons in known disease-associated genes may not have enough coverage to make a sequence cell. Analytic specificity may also be weak with lower depth of coverage, requiring additional Sanger testing to avoid false-positive variant calls.Initial comparisons of WES capture kits showed the capture of approximately 80% of the coding sequence regions at a minimal coverage of 20x [43]. This initial deficiency in sequence coverage for a relatively large amount of the exome has incited clinical laboratories to develop TGP, or custom exome captures to achieve good captures, especially for known genetic disorders [44]. The current estimates of coverage established from whole exome capture and sequencing are 90%–95% at >20x, such as low-coverage regions including target capture, repetitive and GC- or AT -rich regions, copy number variations, and structural variations posing calls for complete capture [45-47]. Higher coverage facilitates more precise differentiation between sequencing errors and real mutations. Cost reduction makes it possible to expand the number of cases to be sequenced, allowing large population-based comparisons.
- Whole-genome sequencing
- Whole-genome sequencing
In a well-planned cohort of children with severe ID, pathogenic CNVs account for 12% of patients; WES identifies the causative genetic mutation in a further 27% of patients. Despite these extensive tests, the etiology of severe ID cases (61%) remains undiagnosed [48]. Therefore, more extensive molecular diagnostic tests are required. WGS enabled sequencing of the entire 3 billion bases of the human genome, including both coding and noncoding DNA. WGS is mostly used in clinical research or in those patients who have been broadly investigated but remain unexplained. WGS currently is the most comprehensive genetic test, but its widespread application to patient diagnostics has been restricted by challenges in processing sequencing data, the test’s unknown diagnostic potential, and relatively high cost compared with other NGS approaches [49,50].WGS is a potential single test that can identify nearly all forms of genomic variation in an unbiased manner, and increasing evidence supports its utility in clinical diagnosis in gene discovery [21,48,51]. Several reports have highlighted the benefits of WGS in mutation detection and WGS analyses of pediatric populations identified clinically relevant variants in approximately 40% of patients with autism and ~60% of those with ID [48,52]. WGS can be used as a single genetic test to accurately identify and defined the extensive spectrum of genetic variation, supporting a genetic diagnosis in the majority of children with severe ID [48,53-55]. WGS as a primary clinical test provided a higher diagnostic success rate than traditional genetic testing in a clinically heterogeneous cohort [56]. Although WGS can be an option, currently it is not widely applied in clinical practice [50].Overall, bioinformatics tools for WGS are less developed than those available for WES or targeted panel sequencing. The cost of WGS is higher than that of WES, partly because of the cost of data handling and analysis. Sequencing throughput is consistently increasing at a stable rate or even at a reduced cost, which also allows for higher sequence coverage for WGS. Enhanced algorithms are being developed for analysis and interpretation of NGS data. The advantage of WGS over other NGS approaches is that patient’s comprehensive genomes are more precise. Advancements in sequence coverage, variant calls, CNV detection, sequencing alignment, and predictions of pathogenicity, are allowing for identifying increasing numbers of new genes, and WGS might improve the diagnostic yield of genomic testing. WGS facilitates analysis of single nucleotide variants, indels, structural variations and CNVs in coding and noncoding regions of the genome. Although WGS provides the most comprehensive data, it is not cost-effective method to find rare variants. WGS also produces large amounts of data, which makes analysis especially complex and resource intensive. To date, WGS has mostly displayed in a lack of coverage of a number of loci also in the reference genome.
- Counseling for genetic tests
- Counseling for genetic tests
Compared with classical genetic counseling, further consideration and reconsidering of genetic counseling principles are required in the territory of NGS. NGS shows variants relevant to a range of disease regardless of the conventional classification of medical specialties. Pretest and posttest counseling with well-informed consent from the patients and their parents for performing WES or WGS is important and should include a discussion of the types of incidental findings that may be found, and whether or not results will be disclosed [57]. Pediatric neurologists should receive additional educational training for genomics, including NGS technology, in order to provide adequate genetic counseling services. In the clinical setting, the useful databases include ExAC, The gnomAD (Genome Aggregation Database), DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources), the MME (Matchmaker Exchange), DGV (Database of Genomic Variants), ClinVar, and dbVar (Database of Genomic Structural Variation).
- Discussion and recommendations
- Discussion and recommendations
Diagnostic approaches have evolved continuously in pediatric neurology clinics with a focus on Mendelian-inherited “diagnostic odyssey” cases that require multiple rounds of diverse testing [32,58]. NGS is a useful tool for genetic testing for and diagnosis of heterogeneous disorders in clinical settings, especially in pediatric populations in whom all the clinical features may not yet be clear. NGS technology can be personalized as a diagnostic tool for rare diseases and serves as an alternative to invasive diagnostic modalities. It can also be used to predict future health conditions, estimate the risk of recurrence in parents and other family members, and accurately identify other family members carrying the mutations for appropriate interventions. TGP, WES, and WGS are applied today in medical practice with children with DD/ID [21,59]. Before performing NGS, it is essential to determine that the individual gene is covered and that the testing is adequate.Standard approaches are often entail time-consuming algorithmic involving CMA and TGP, which can be costly and inconclusive. Clinical guidelines are necessary for optimizing patient selection for NGS and we suggested a clinical workflow (Fig. 3). However, clinicians modify specific tests for differing various phenotypes and different setting of equipment in clinical era. TGP or WES in children with DD/ID will be the most efficient clinical approaches to early diagnosis. In cases of genetic heterogeneity of DD/ID, TGP focused on a wide range of differential diagnosis can reduce the cost and result in appropriate outcomes. WES or WGS should also be considered when clinical features are nonspecific and affect multisystemic symptoms, both of which make it difficult to select a particular panel. NGS is a candidate for first-tier genetic tests in certain neurologic disorders such as Charcot-Maire Tooth disease, syndromic epilepsy, or dystonia [60]. WES and WGS involving total sequencing of the genome or the protein-coding regions of the genome can replace targeted panel sequencing. WGS and WES represent a comprehensive testing platform with the potential to simplify genetic assessment; however, limited comparative data are available to guide its clinical application. A few technical limitations are as follows: WES cannot be used to detect mutations in noncoding regions of the human genome and neither WES nor WGS covers repetitive regions. Most WES/WGS cannot cover platforms accurately and find CNVs [61]. Microdeletion/duplication could not be reliably detected from TGP or WES. Current NGS tests miss diseases associated with trinucleotide expansion such as fragile X syndrome. Clinician has to understand of mutation spectrum, various genetic test methods, and diagnostic sensitivity and specificity. Sequencing depth affects the acute analysis of genome fragment and detection of mutations (e.g., potential false negative data). While 30xWGS was once the average coverage for genome sequencing in the former days, investigators on recent simulations recommend that sequencing coverage be extended [62,63]. There is ample room for further technical advancements considering the unequal depth of coverage in the exome, differences in coverage, and mapping problems. Selecting the appropriate genetic testing method should be based on cost-effectiveness, depth of coverage, and optimal timing to guide clinician decision-making [36,41,64,65].WES and WGS not only give a great opportunity to acquire genomic contributions to disease, but also have the potential to find genetic information that may not relate to the patient’s presenting morbidities, including variants associated with other health problems, which may or may not be medically actionable and incidental [66]. The use of WES, WGS, or any type of sequencing is considered as a legal and ethical issues related to data security as well as to how and when to make share findings with the patient or family, and of data security, so these areas need to be approached [67]. WES and WGS identify Incidental findings in 0.5%–3.5% of individuals tested although this will improve as our understanding of genetic causes of disease advances [68,69]. Due to the difficulties in analyzing and interpreting the large amounts of raw data, the widespread application of WES or WGS is not straightforward [70].Diagnosis yields defined as the assured association of a genotype with the clinical phenotype range from 20% to 60% according to multiple factors, including specificity of the clinical manifestation, genetic heterogeneity of the disorder, patient enrollment, evidence of de novo incident of causal variants, and date of publication (Table 1) [71,72]. Approaches to finding novel disease-causing genes have profitably applied de novo trio analysis to compare the proband’s genome with the parental genomes with diagnostic yields of 15% to 40% and an additional 10% to 20% utilizing homozygosity mapping or biallelic variant analysis approach [38,48,73,74]. Cost-effective, trio-based exome sequencing has aided in determining de novo alterations in DD/ID incidence. Family-based sequencing analysis is important for identifying pathologic genes for Mendelian disorders and rare associated variants [75]. In a recent report, exome reanalysis 12 months after the primary interpretation showed additional diagnoses, making it a cost-saving diagnostic approach [76]. With rapid increases in the amount of available genetic information, systemic and retrospective reanalysis of raw genomic data will result in better diagnostic yields [77].
- Conclusion
- Conclusion
NGS offers expanded the diagnostic options in children with undiagnosed DD/ID as well as investigation into the pathogenesis of these disorders. Genetic diagnosis plays an important role by assisting patients and families with (1) preventing of inappropriate managements and additional invasive tests such as muscle biopsy, (2) family genetic counseling, and (3) proper therapy and information pertaining to disease and prognosis. Patient sequencing will become routine in clinical settings in developed countries, specifically as sequencing throughout will further make increment at reduced cost [78]. Decreasing in the cost of NGS will bring on widespread use of family-based trio testing by WES/WGS as second-tier test for DD/ID, leading to a higher diagnostic yield [79,80]. Further studies (including ethical, clinical utility, and cost-benefit analysis) are necessary before the routine application of NGS in clinical praxis.Not notably, since 2001 NGS had identified a large number of genetic etiologies. From 2007 to 2013, the diagnosis of Online Mendelian Inheritance of Man introduced new Mendelian disorders that expanded database by 87%, with many them associated with pediatric neurologic diseases [81,82], and new discovery still in progress day by day. Collaboration among pediatric neurologists and laboratory geneticists is recommended in the planning, implementation, and development of genetic testing. With the wide application of NGS, we also should know that NGS is not “all-powerful.” NGS is not suitable for screening for nucleotide repeat expansion disease or detecting large deletions or duplications. A persistent progression of diagnostic genomic testing by NGS is anticipated within the years to come. Comprehensive mapping of the whole genome in conjunction with further studies investigating their functional elements, will be important in the future to comprehend the function of noncoding DNA regions in DD/ID. International collaboration will become increasingly important in genetic researches on children with DD/ID.
- Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Fig. 1.
Algorithm for a stepwise approach in children with developmental delay/intellectual disability [10,11,83-85]. Nowadays, second-tier genetic tests using NGS can be considered before other invasive or expensive evaluation (dotted arrows). CMA, chromosomal microarray; FMR1, fragile X mental retardation1; NGS; next-generation sequencing.

Fig. 2.
Comparison of targeted gene panel, whole exome sequencing, and whole genome sequencing approaches.
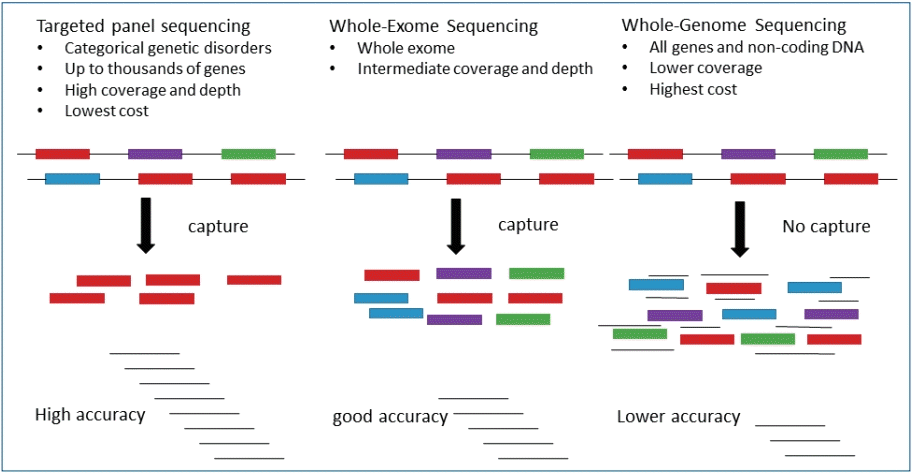
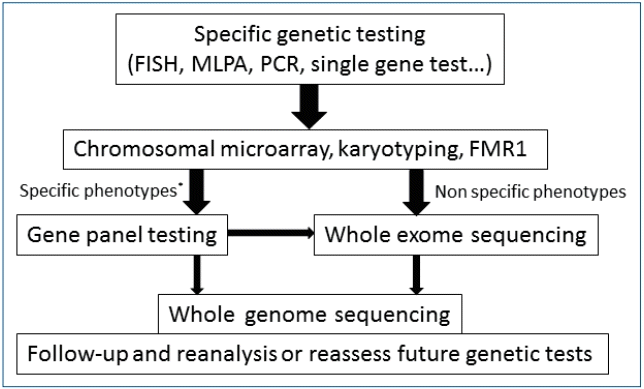
Table 1.
Diagnostic yield of developmental delay and/or intellectual disability using next-generation sequencing
| Method | Study | No. of subjects | Diagnostic yield (%) | Remarks |
|---|---|---|---|---|
| Gene panel | Pekeles et al. [86] (2019) | 48 | 21 | |
| Gieldon et al. [88] (2018) | 106 | 30 | Developmental disorders | |
| Han et al. [89] (2018) | 35 | 29 | ||
| Reid et al. [90] (2016) | 30 | 89 | Neurometabolic phenotypes | |
| Grozeva et al. [94] (2015) | 986 | 11 | Moderate to severe ID | |
| Brett et al. [91] (2014) | 8 | 25 | ID, congenital anomalies, and/or ASD | |
| De Ligt et al. [38] (2012) | 100 | 53 | Severe ID | |
| WES | Bowling et al. [87] (2017) | 127 | 30 | |
| Kuperberg et al. [92] (2016) | 57 | 49 | Pediatric neurology patientsa) | |
| Srivastava et al. [93] (2014) | 78 | 41 | Pediatric neurology patientsb) | |
| Gilissen et al. [48] (2014) | 100 | 27 | Severe ID | |
| Rauch et al. [39] (2012) | 51 | 45-55 | Severe ID | |
| WGS | Bowling et al. [87] (2017) | 244 | 22 | |
| Gilissen et al. [48] (2014) | 50 | 42 | Severe ID, previous negative results in WES | |
| Jiang et al. [52] (2013) | 32 Families | 50 | ASD |
- References
- 1. Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE, et al. The epidemiology of autism spectrum disorders. Annu Rev Public Health 2007;28:235–58.
[Article] [PubMed]2. Srour M, Shevell M. Genetics and the investigation of developmental delay/intellectual disability. Arch Dis Child 2014;99:386–9.
[Article] [PubMed]3. Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003;60:367–80.
[Article] [PubMed]4. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977;74:5463–7.
[Article] [PubMed] [PMC]5. Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet 2010;11:31–46.
[Article] [PubMed]6. Slatko BE, Gardner AF, Ausubel FM. Overview of next-generation sequencing technologies. Curr Protoc Mol Biol 2018;122:e59.
[Article] [PubMed] [PMC]7. Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 2013;15:733–47.
[Article] [PubMed] [PMC]8. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–64.
[Article] [PubMed] [PMC]9. O'Byrne JJ, Lynch SA, Treacy EP, King MD, Betts DR, Mayne PD, et al. Unexplained developmental delay/learning disability: guidelines for best practice protocol for first line assessment and genetic/metabolic/radiological investigations. Ir J Med Sci 2016;185:241–8.
[Article] [PubMed]10. Li Y, Anderson LA, Ginns EI, Devlin JJ. Cost effectiveness of karyotyping, chromosomal microarray analysis, and targeted next-generation sequencing of patients with unexplained global developmental delay or intellectual disability. Mol Diagn Ther 2018;22:129–38.
[Article] [PubMed]11. Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med 2014;16:176–82.
[Article] [PubMed]12. van Karnebeek CD, Scheper FY, Abeling NG, Alders M, Barth PG, Hoovers JM, et al. Etiology of mental retardation in children referred to a tertiary care center: a prospective study. Am J Ment Retard 2005;110:253–67.
[Article] [PubMed]13. Hunter AG. Outcome of the routine assessment of patients with mental retardation in a genetics clinic. Am J Med Genet 2000;90:60–8.
[Article] [PubMed]14. Schaefer GB, Lutz RE. Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genet Med 2006;8:549–56.
[Article] [PubMed]15. Moog U. The outcome of diagnostic studies on the etiology of mental retardation: considerations on the classification of the causes. Am J Med Genet A 2005;137:228–31.
[Article] [PubMed]16. Mithyantha R, Kneen R, McCann E, Gladstone M. Current evidencebased recommendations on investigating children with global developmental delay. Arch Dis Child 2017;102:1071–6.
[Article] [PubMed] [PMC]17. Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 2013;14:681–91.
[Article] [PubMed]18. Stavropoulos DJ, Merico D, Jobling R, Bowdin S, Monfared N, Thiruvahindrapuram B, et al. Whole genome sequencing expands diagnostic utility and improves clinical management in pediatric medicine. NPJ Genom Med 2016;1.
[Article]19. Proost D, Vandeweyer G, Meester JA, Salemink S, Kempers M, Ingram C, et al. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat 2015;36:808–14.
[Article] [PubMed]20. Vasudevan P, Suri M. A clinical approach to developmental delay and intellectual disability. Clin Med (Lond) 2017;17:558–61.
[Article] [PubMed] [PMC]21. Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014;6:265ra168
[Article] [PubMed] [PMC]22. Ankala A, da Silva C, Gualandi F, Ferlini A, Bean LJ, Collins C, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol 2015;77:206–14.
[Article] [PubMed]23. Saudi Mendeliome Group. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol 2015;16:134
[Article] [PubMed] [PMC]24. Lim EC, Brett M, Lai AH, Lee SP, Tan ES, Jamuar SS, et al. Next-generation sequencing using a pre-designed gene panel for the molecular diagnosis of congenital disorders in pediatric patients. Hum Genomics 2015;9:33
[Article] [PubMed] [PMC]25. de Koning TJ, Jongbloed JD, Sikkema-Raddatz B, Sinke RJ. Targeted next-generation sequencing panels for monogenetic disorders in clinical diagnostics: the opportunities and challenges. Expert Rev Mol Diagn 2015;15:61–70.
[Article] [PubMed]26. Wang W, Wang C, Dawson DB, Thorland EC, Lundquist PA, Eckloff BW, et al. Target-enrichment sequencing and copy number evaluation in inherited polyneuropathy. Neurology 2016;86:1762–71.
[Article] [PubMed] [PMC]27. Klein CJ, Middha S, Duan X, Wu Y, Litchy WJ, Gu W, et al. Application of whole exome sequencing in undiagnosed inherited polyneuropathies. J Neurol Neurosurg Psychiatry 2014;85:1265–72.
[Article] [PubMed]28. Wang J, Gotway G, Pascual JM, Park JY. Diagnostic yield of clinical nextgeneration sequencing panels for epilepsy. JAMA Neurol 2014;71:650–1.
[Article] [PubMed]29. Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med 2015;17:253–61.
[Article] [PubMed]30. Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed 2013;98:236–8.
[Article] [PubMed] [PMC]31. Winkler EC, Wiemann S. Findings made in gene panel to whole genome sequencing: data, knowledge, ethics - and consequences? Expert Rev Mol Diagn 2016;16:1259–70.
[Article] [PubMed]32. Klein CJ, Foroud TM. Neurology individualized medicine: when to use next-generation sequencing panels. Mayo Clin Proc 2017;92:292–305.
[Article] [PubMed]33. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011;12:745–55.
[Article] [PubMed]34. Need AC, Shashi V, Schoch K, Petrovski S, Goldstein DB. The importance of dynamic re-analysis in diagnostic whole exome sequencing. J Med Genet 2017;54:155–6.
[Article] [PubMed]35. Bowdin S, Gilbert A, Bedoukian E, Carew C, Adam MP, Belmont J, et al. Recommendations for the integration of genomics into clinical practice. Genet Med 2016;18:1075–84.
[Article] [PubMed] [PMC]36. Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015;17:578–86.
[Article] [PubMed]37. Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, Guzman E, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med 2014;16:922–31.
[Article] [PubMed]38. De Ligt J, Willemsen MH, Van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–9.
[Article] [PubMed]39. Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012;380:1674–82.
[Article] [PubMed]40. Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, Weiss ME, Koster J, Marais A, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 2017;25:176–82.
[Article] [PubMed]41. Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014;312:1880–7.
[Article] [PubMed] [PMC]42. Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med 2017;19:209–14.
[Article] [PubMed]43. Parla JS, Iossifov I, Grabill I, Spector MS, Kramer M, McCombie WR. A comparative analysis of exome capture. Genome Biol 2011;12:R97
[Article] [PubMed] [PMC]44. Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of nextgeneration sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 2015;17:444–51.
[Article] [PubMed]45. Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol 2011;29:908–14.
[Article] [PubMed] [PMC]46. Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med 2014;370:2418–25.
[Article] [PubMed]47. Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol 2011;12:R68
[Article] [PubMed] [PMC]48. Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014;511:344–7.
[Article] [PubMed]49. Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 2010;362:1181–91.
[Article] [PubMed] [PMC]50. Meynert AM, Ansari M, FitzPatrick DR, Taylor MS. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 2014;15:247
[Article] [PubMed] [PMC]51. Pang AW, Macdonald JR, Yuen RK, Hayes VM, Scherer SW. Performance of high-throughput sequencing for the discovery of genetic variation across the complete size spectrum. G3 (Bethesda) 2014;4:63–5.
[Article] [PubMed]52. Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by wholegenome sequencing. Am J Hum Genet 2013;93:249–63.
[Article] [PubMed] [PMC]53. Glockle N, Kohl S, Mohr J, Scheurenbrand T, Sprecher A, Weisschuh N, et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet 2014;22:99–104.
[Article] [PubMed]54. Wei Q, Zhu H, Qian X, Chen Z, Yao J, Lu Y, et al. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. J Transl Med 2014;12:311
[Article] [PubMed] [PMC]55. Zhao L, Wang F, Wang H, Li Y, Alexander S, Wang K, et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum Genet 2015;134:217–30.
[Article] [PubMed]56. Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435–43.
[Article] [PubMed]57. Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol 2012;19:173–80.
[Article] [PubMed]58. Foo JN, Liu JJ, Tan EK. Whole-genome and whole-exome sequencing in neurological diseases. Nat Rev Neurol 2012;8:508–17.
[Article] [PubMed]59. Green ED, Guyer MS. Charting a course for genomic medicine from base pairs to bedside. Nature 2011;470:204–13.
[Article] [PubMed]60. Huang Y, Yu S, Wu Z, Tang B. Genetics of hereditary neurological disorders in children. Transl Pediatr 2014;3:108–19.
[PubMed] [PMC]61. Bras J, Guerreiro R, Hardy J. Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev Neurosci 2012;13:453–64.
[Article] [PubMed]62. Wetterstrand KA. DNA sequencing costs: data [Internet]. Bethesda (MD), National Human Genome Research Institute. 2013;[cited 2010 Mar 1]. Available from: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data.63. McGinn S, Gut IG. DNA sequencing - spanning the generations. N Biotechnol 2013;30:366–72.
[Article] [PubMed]64. Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312:1870–9.
[Article] [PubMed] [PMC]65. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature 2001;409:860–921.
[Article] [PubMed]66. Jarvik GP, Amendola LM, Berg JS, Brothers K, Clayton EW, Chung W, et al. Return of genomic results to research participants: the floor, the ceiling, and the choices in between. Am J Hum Genet 2014;94:818–26.
[Article] [PubMed] [PMC]67. Townsend A, Adam S, Birch PH, Lohn Z, Rousseau F, Friedman JM. "I want to know what's in Pandora's Box": comparing stakeholder perspectives on incidental findings in clinical whole genomic sequencing. Am J Med Genet A 2012;158a:2519–25.
[Article] [PubMed]68. Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res 2015;25:305–15.
[Article] [PubMed] [PMC]69. Tabor HK, Auer PL, Jamal SM, Chong JX, Yu JH, Gordon AS, et al. Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results. Am J Hum Genet 2014;95:183–93.
[Article] [PubMed] [PMC]70. Robinson PN, Krawitz P, Mundlos S. Strategies for exome and genome sequence data analysis in disease-gene discovery projects. Clin Genet 2011;80:127–32.
[Article] [PubMed]71. Taylor JC, Martin HC, Lise S, Broxholme J, Cazier JB, Rimmer A, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 2015;47:717–26.
[Article] [PubMed] [PMC]72. Smith HS, Swint JM, Lalani SR, Yamal JM, de Oliveira Otto MC, Castellanos S, et al. Clinical application of genome and exome sequencing as a diagnostic tool for pediatric patients: a scoping review of the literature. Genet Med 2019;21:3–16.
[Article] [PubMed]73. Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genetics 2014;10:e1004772.
[Article] [PubMed] [PMC]74. Musante L, Ropers HH. Genetics of recessive cognitive disorders. Trends Genet 2014;30:32–9.
[Article] [PubMed]75. Yang Z, Thomas DC. Two-stage family-based designs for sequencing studies. BMC Proc 2014;8:S32
[Article] [PubMed] [PMC]76. Ewans LJ, Schofield D, Shrestha R, Zhu Y, Gayevskiy V, Ying K, et al. Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet Med 2018;20:1564–74.
[Article] [PubMed]77. Smith ED, Radtke K, Rossi M, Shinde DN, Darabi S, El-Khechen D, et al. Classification of genes: standardized clinical validity assessment of genedisease associations aids diagnostic exome analysis and reclassifications. Hum Mutat 2017;38:600–8.
[Article] [PubMed] [PMC]78. Mandl KD, Kohane IS. Time for a patient-driven health information economy? N Engl J Med 2016;374:205–8.
[Article] [PubMed]79. Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 2016;17:9–18.
[Article] [PubMed]80. Hayeems RZ, Babul-Hirji R, Hoang N, Weksberg R, Shuman C. Parents' experience with pediatric microarray: transferrable lessons in the era of genomic counseling. J Genet Couns 2016;25:298–304.
[Article] [PubMed]81. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM. org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res 2015;43(Database issue):D789–98.82. McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet 2007;80:588–604.
[Article] [PubMed] [PMC]83. Han JY, Jang W, Park J, Kim M, Kim Y, Lee IG. Diagnostic approach with genetic tests for global developmental delay and/or intellectual disability: Single tertiary center experience. Ann Hum Genet 2019;83:115–23.
[Article] [PubMed]84. Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol 2012;19:173–80.
[Article] [PubMed]85. Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2011;77:1629–35.
[Article] [PubMed]86. Pekeles H, Accogli A, Boudrahem-Addour N, Russell L, Parente F, Srour M. Diagnostic yield of intellectual disability gene panels. Pediatr Neurol 2019;92:32–6.
[Article] [PubMed]87. Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med 2017;9:43
[Article] [PubMed] [PMC]88. Gieldon L, Mackenroth L, Kahlert AK, Lemke JR, Porrmann J, Schallner J, et al. Diagnostic value of partial exome sequencing in developmental disorders. PLoS One 2018;13:e0201041.
[Article] [PubMed] [PMC]89. Han JY, Jang JH, Park J, Lee IG. Targeted next-generation sequencing of Korean patients with developmental delay and/or intellectual disability. Front Pediatr 2018;6:391
[Article] [PubMed] [PMC]90. Reid ES, Papandreou A, Drury S, Boustred C, Yue WW, Wedatilake Y, et al. Advantages and pitfalls of an extended gene panel for investigating complex neurometabolic phenotypes. Brain 2016;139:2844–54.
[Article] [PubMed] [PMC]91. Brett M, McPherson J, Zang ZJ, Lai A, Tan ES, Ng I, et al. Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. PLoS One 2014;9:e93409.
[Article] [PubMed] [PMC]92. Kuperberg M, Lev D, Blumkin L, Zerem A, Ginsberg M, Linder I, et al. Utility of whole exome sequencing for genetic diagnosis of previously undiagnosed pediatric neurology patients. J Child Neurol 2016;31:1534–9.
[Article] [PubMed]