



Introduction
Neonatal ichthyosis-sclerosing cholangitis (NISCH) syndrome, also known as ichthyosis, leukocyte vacuoles, alopecia, and sclerosing cholangitis (ILVASC), is a rare autosomal recessive disorder characterized by a combination of ichthyosis, sclerosing cholangitis, and dental and hair changes. The syndrome is caused by pathogenic variations in the CLDN1 gene (chromosome 3q27-q28) [1]. NISCH syndrome was first described in patients of Moroccan ancestry by Baala et al. in 2002 [2]. It has a characteristic presentation at birth with generalized ichthyosis and neonatal cholestasis. Liver involvement varies, ranging from transient neonatal cholestasis to end-stage liver disease. Its hallmark dermatological manifestations include ichthyosis, hair changes such as alopecia and hypotrichosis, and dental anomalies such as enamel hypoplasia. Other differential diagnoses of neonatal cholestasis with ichthyosis include arthrogryposis-renal dysfunction-cholestasis syndrome, MEDNIK (mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, keratoderma) syndrome. As it is a rare syndrome caused by pathogenic variations in a single gene (CLDN1), increased knowledge of NISCH syndrome will not only expedite early diagnosis by targeted genetic analysis but also avoid misdiagnosis. Here we report a novel genetic mutation and review 37 genetically confirmed cases published to date. This review highlights the variable disease phenotypes and multisystem involvement and underscores the importance of early genetic diagnosis for the multidisciplinary management of NISCH syndrome
Methods
We searched for original articles, case series, and case reports describing clinical profiles of NISCH syndrome on PubMed using the keywords “NISCH syndrome” or “ILVASC syndrome” or “Ichthyosis” AND “Sclerosing cholangitis” for articles published between January 2002 and December 2024. The relevant literature was retrieved and independently assessed for eligibility by 2 authors and then reviewed by the remaining authors. We identified 32 articles, of which 21 included clinical descriptions of 37 genetically confirmed cases. We summarized the clinical findings, including age at presentation, hepatic and dermatological involvement, affected ethnicities and their mutations, diagnostic work-up findings, and patient outcomes. Written informed consent was taken from the parents for anonymous publication for the patient’s clinical details and photographs of the case report. Here we present a case report, followed by a detailed review of NISCH syndrome including all cases published through December 2024.
Case report
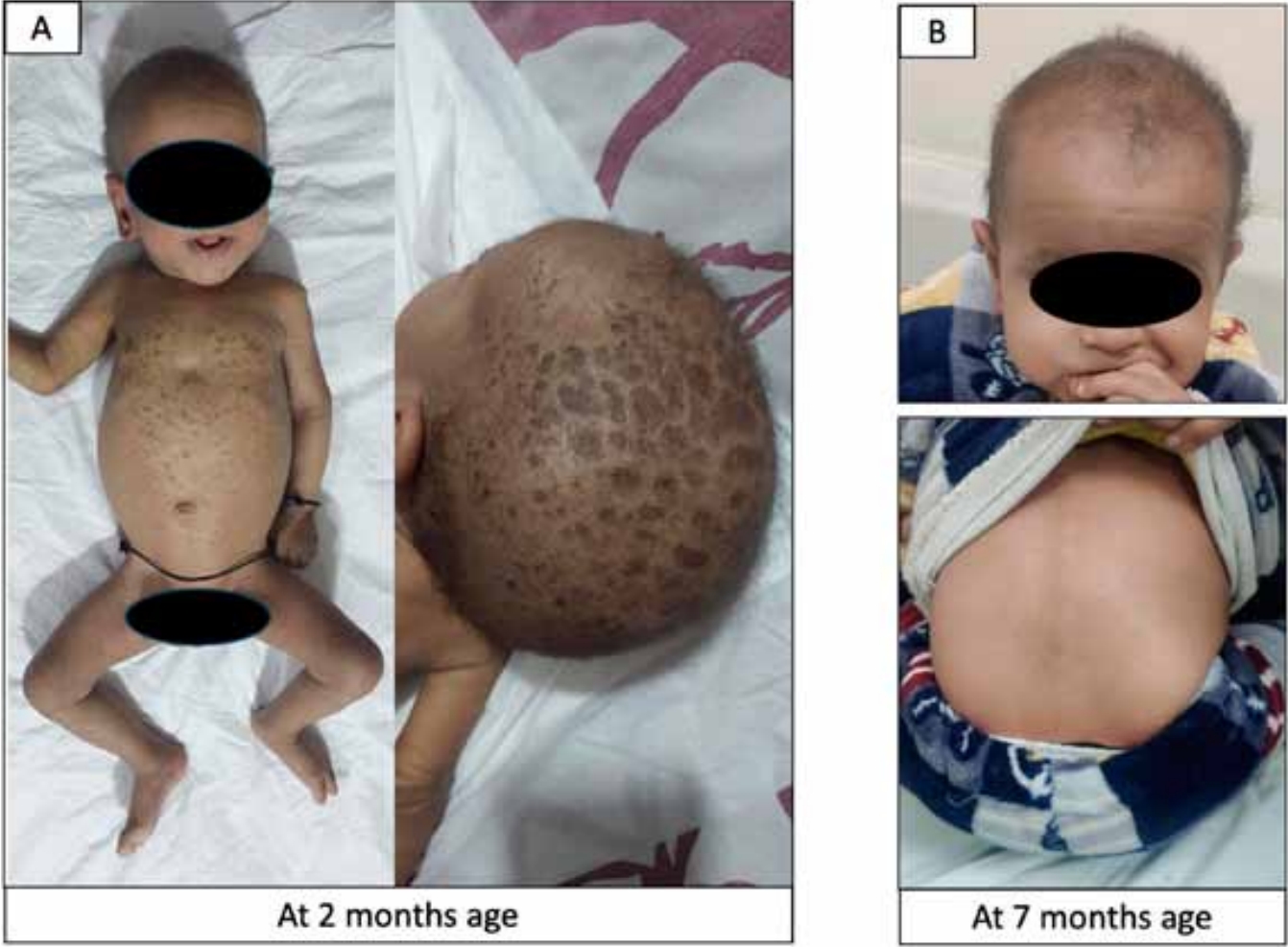
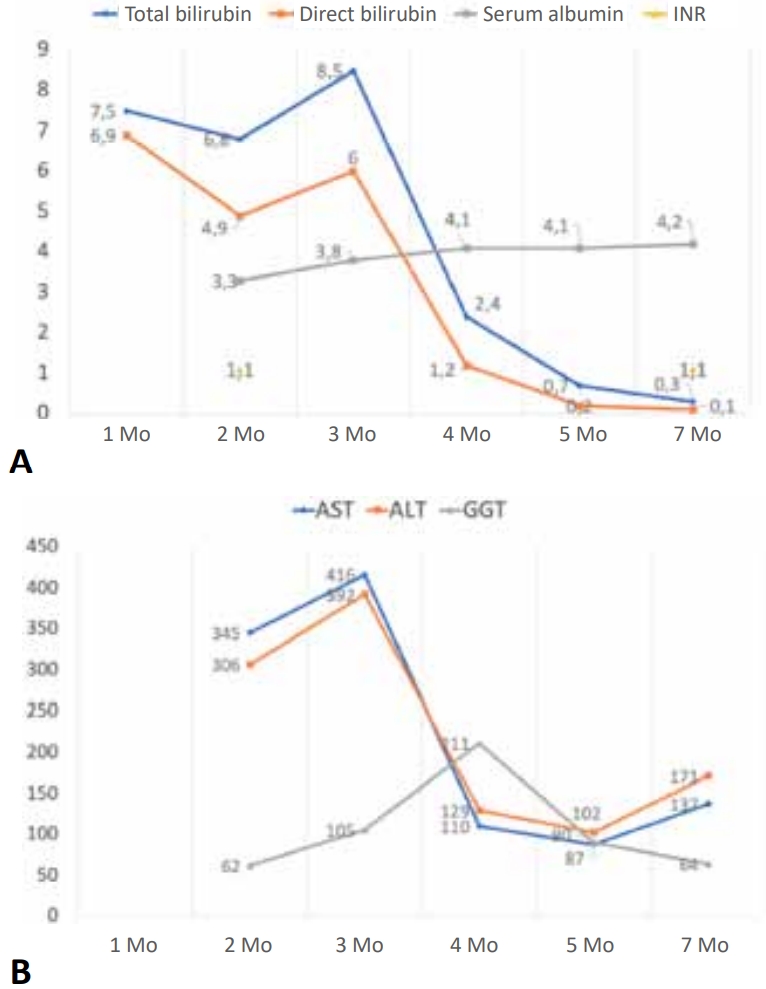
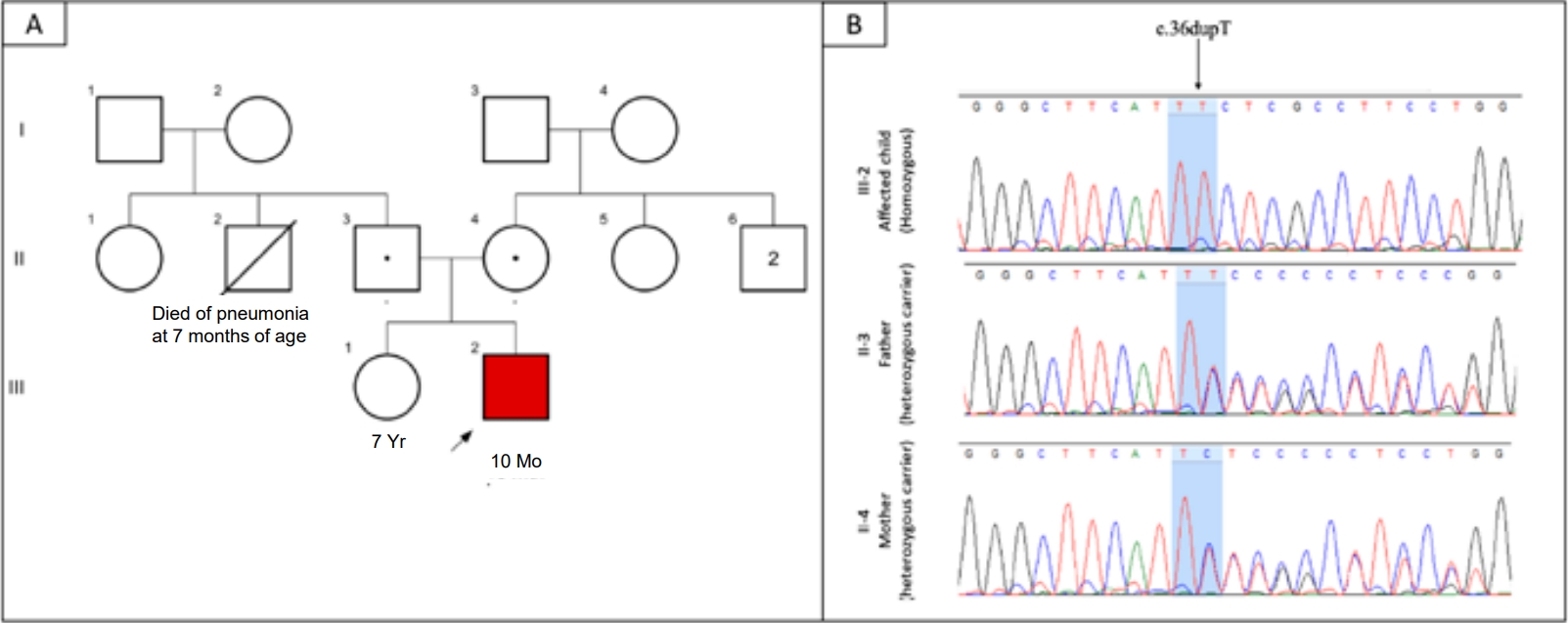
A 2-month-old boy born to nonconsanguineous parents (term delivery, no perinatal complications) presented with jaundice, dark urine, pigmented stools, and generalized ichthyosis since the first week of life. He weighed 4.3 kg (-1.92 standard deviation [SD]) and measured 61 cm (-1.21 SD). A physical examination revealed icterus, generalized ichthyosis, and sparse hair and eyebrows (Fig. 1A). An abdominal examination revealed soft hepatomegaly (palpable 4 cm below the right costal margin) and splenomegaly (2 cm). Laboratory investigations revealed conjugated hyperbilirubinemia with preserved synthetic liver function (Fig. 2). The hemogram was normal. Abdominal ultrasonography revealed a good-sized gallbladder (2.5 cm) and a normal common bile duct (1.6 mm). An ophthalmological examination revealed no evidence of uveal synechia or cataracts. We suspected NISCH syndrome based on the presence of neonatal cholestasis with generalized ichthyosis and sparse hair. Whole-exome sequencing (WES) revealed a homozygous variant in exon 1 of the CLDN1 gene: (NM_021101.5) c.36dupT (p.Leu13SerfsTer56). This variant was classified as likely pathogenic based on American College of Medical Genetics and Genomics criteria. Sanger sequencing revealed that both parents were heterozygous asymptomatic carriers of the variant (Fig. 3).
The infant was treated with vitamins D (800 IU/day) and E (200 IU/day), ursodeoxycholic acid (20 mg/kg/day), and multivitamins. Medium-chain triglyceride-based oil was added for caloric supplementation. Urea-based emollient creams were used to treat the ichthyosis. By the 7-month follow-up, the ichthyosis and jaundice had resolved completely, although mild pruritus persisted (Fig. 1B).
Discussion
Our patient presented with cholestasis, hepatosplenomegaly, generalized ichthyosis, sparse hair, and elevated transaminase levels. WES identified a novel variant in the CLDN1 gene (c.36dupT, p. Leu13SerfsTer56, exon 1) that resulted in a premature termination codon, leading to a truncated protein product. This clinical picture was similar to the 37 globally reported cases among patients of various ethnicities (Table 1) [1-21]. In the literature review, the median age at diagnosis was 60 months (range, 1–636 months), and consanguinity was reported in 56.7% of cases (21 of 37). At presentation, ichthyosis was universal (37 of 37 [100%]), followed by jaundice in 70.2% (26 of 37), pruritus in 38.2% (13 of 34), hepatomegaly in 43.3% (13 of 30), and splenomegaly in 23.8% (5 of 21) of the patients (Table 1). Associated features included hypotrichosis/alopecia (30 of 36 [83.3%]), oligodontia/hypodontia (11 of 28 [39.3%]), enamel hypoplasia (16 of 27 [59.2%]), and uveal synechiae (1 patient). Portal hypertension (7 of 35 [20%]) and mental retardation (3 of 21 [14.2%]) were rare. A peripheral blood smear showed vacuolated eosinophils in 30% (3 of 10) of patients. All patients were homozygous for pathogenic variants of the CLDN1 gene, the most common variant was exon 1, c.200_201delTT (16 of 37 [43.2%]), followed by c.242G>A (9 of 37 [24.3%]), and the parents were typically asymptomatic.
Percutaneous cholangiography performed in 5 patients revealed sclerosing cholangitis in 3 (at 5–6 years of age) and hypoplastic bile ducts in 2 (age unknown). Magnetic resonance cholangiopancreatography was performed in one patient, which was normal [2,4,9]. A liver biopsy performed in 14 patients revealed fibrosis and bile duct proliferation in 50% (7 of 14) as well as ductopenia in 21.4% (3 of 14) [1,2,4,5,8,9,12,14,18,21]. On liver biopsy, the typical ‘‘onion-skin’’ appearance of periductal fibrosis was not seen as expected considering the age of the patients [22]. Ductopenia, a well-documented histopathological feature of sclerosing cholangitis, is most commonly observed in stage 3 of the disease according to the Ludwig classification [23]. Nagtzaam et al. [4] reported 2 cases of bile duct paucity without any significant fibrosis, suggesting that it may be a primary phenomenon due to abnormal bile duct development rather than a sequela. On the contrary, Paganelli et al. [5] showed ductopenia in the explant liver of a patient at 1 year of age that was not present at initial presentation. Thus, long-term follow-up and explant liver biopsy may aid our understanding of the biliary pathology in NISCH syndrome. In our patient, a liver biopsy was deferred since the liver function tests showed improvement. Another difficulty associated with this condition is the close resemblance of its clinical features to those of biliary atresia, which may lead to a misdiagnosis. Four cases in the literature were initially misdiagnosed as biliary atresia; 3 of these patients underwent the Kasai procedure within the first 2 months of life [5,16,18,21]. These patients subsequently progressed to chronic liver disease: 3 of 4 underwent liver transplantation at the ages of 1 and 8 years (age unavailable for the third) [5,16,18,21]. This underscores the importance of diagnostic accuracy to avoid the Kasai procedure.
Ichthyosis, a universal finding secondary to defects in the epidermal barrier, cannot be corrected by liver transplantation [5,9,16,18]. In contrast, Baala et al. [2] reported the case of a patient in whom complete regression of ichthyosis and alopecia was noted post-transplant while on immunosuppression with tacrolimus, a finding that remains unexplained. A receding frontal hairline, alopecia, hypotrichosis, and curly hair are other characteristic dermatological findings (Table 1).
The CLDN1 gene encodes claudin-1, a key protein in tight junctions found in keratinocytes, hepatocytes, and cholangiocytes [2,11,24,25]. Defective tight junctions result in loss of the epidermal barrier function. In the liver, paracellular leakage of bile from the biliary canaliculi leads to pericholangiocellular inflammation, fibrosis, and bile duct obliteration, resulting in sclerosing cholangitis. Furthermore, claudin-1 is also expressed in extrahepatic bile ducts, and tight-junction dysfunction has been associated with abnormal bile duct development [11,24,25]. Claudin-1 is also expressed in the tight junctions of developing enamel-secreting cells (ameloblasts) and hair follicles, which explains the enamel defects and abnormal hair phenotype present in these patients. The pathogenesis of pruritus in NISCH syndrome is likely related to epithelial barrier dysfunction (bile acid levels were normal in one case report) [5,9,19].
Differential diagnoses for infants presenting with generalized ichthyosis and cholestasis include arthrogryposis, renal dysfunction, cholestasis syndrome; MEDNIK syndrome; and type 2 Gaucher disease (Table 2). In our case, WES performed with full coverage of the above genes did not reveal any significant single nucleotide variants or large copy number variations in the genes associated with the other differential diagnoses.
The natural history and prognosis of NISCH syndrome remain unclear; perhaps, it is underdiagnosed, as only 37 cases have been reported to date. However, its prevalence appears to vary among ethnicities. A literature search showed 12 significant disease-causing variations (missense, n=3; nonsense, n=9) in the CLDN1 gene that included exon 1—c.200_201delTT, the Moroccan founder mutation in 40.5% (15 of 37), followed by c.242G>A variant in 24.3% (9 of 37), c.181C>T, p.Gln61X in the Turkish population (n=3), and c.578C>A, p.Tyr159Ter in the Kurdish (Iranian) population (n=2) and a homozygous deletion in the nucleotide of exon 2, c.358delG, p.Val120fs in the Swiss population (n=1). The remaining variants are shown in Table 1. All patients were homozygous for pathogenic variants. Exon 1 of the CLDN1 gene seems to be a hotspot for mutations, and targeted cost-effective approaches such as Sanger sequencing can be performed if NISCH syndrome is suspected. Ours is the fourth case reported of a patient of Indian ethnicity; all 3 patients with different mutations described previously, had transient neonatal cholestasis with a good prognosis [12,17,19]. The Swiss, Swiss-Turkish, and Turkish mutations reportedly have a better prognosis (regression of cholestasis, n=2; no liver involvement, n=2), in contrast to the Moroccan mutation, which is reportedly associated with variable outcomes ranging from no liver involvement to severe liver disease, making the prognosis unpredictable. Phenotypic variation within the same family is well documented, even within the same mutation, ranging from no liver involvement to advanced liver disease [2,3,4,5,6,18]. The presence of phenotypic variability in the same family complicates the genotype–phenotype relationship. Our patient's family history was noncontributory (Fig. 3A). In the literature review, followup data were available for 34 patients; 8 of them (23.5%) had isolated skin involvement without any liver involvement. Among those with liver disease, bilirubin had normalized in 52.9% (18 of 34) by age 1–29 months. Notably, in 6 studies, 8 patients progressed to chronic liver disease, of whom 5 underwent liver transplantation [2,4,5,16,18,21]. This highlights that NISCH syndrome carries significant morbidity and requires monitoring for the progression of liver disease. It is difficult to determine whether transient neonatal cholestasis responds to ursodeoxycholic acid or it is the natural history of the disease.
The management of NISCH syndrome primarily focuses on symptom management, i.e., the treatment of cholestasis with ursodeoxycholic acid and fat-soluble vitamin supplementation, use of moisturizers and emollients, good hydration for the ichthyosis, and dental coverings with crowns or resins for enamel dysplasia. In severe cases, liver transplantation may be necessary to treat the end-stage liver disease. We recommend 3–6 monthly follow-ups with growth monitoring; skin, hair, and teeth examinations; monitoring of liver and spleen size by ultrasonography; liver function tests; regular cognitive assessments to identify intellectual delays; and ophthalmological examinations for uveal synechiae. Liver stiffness can be measured by using transient elastography to identify portal hypertension. Parental counseling regarding the natural history of the disease, especially regarding complications related to liver disease, is necessary. Genetic counseling and prenatal testing for families with a history of NISCH syndrome (25% risk of recurrence) enables its early diagnosis.
In summary, here we presented a case of NISCH syndrome with transient cholestasis and ichthyosis and highlighted a novel mutation. NISCH syndrome poses diagnostic and therapeutic challenges owing to its clinical heterogeneity and rarity. The specific association of ichthyosis, hypotrichosis, and cholestasis in the context of consanguinity is a key element in guiding the diagnosis. Moreover, its management requires a multidisciplinary approach involving dermatologists, dentists, hepatologists, and geneticists. It is important to note that liver transplantation does not resolve extrahepatic manifestations and should be reserved for patients with end-stage liver disease. To date, most of the literature on NISCH syndrome comprises short case reports and case series; thus, long-term follow-up studies are required to better elucidate the natural history and genetics of this rare condition.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


