




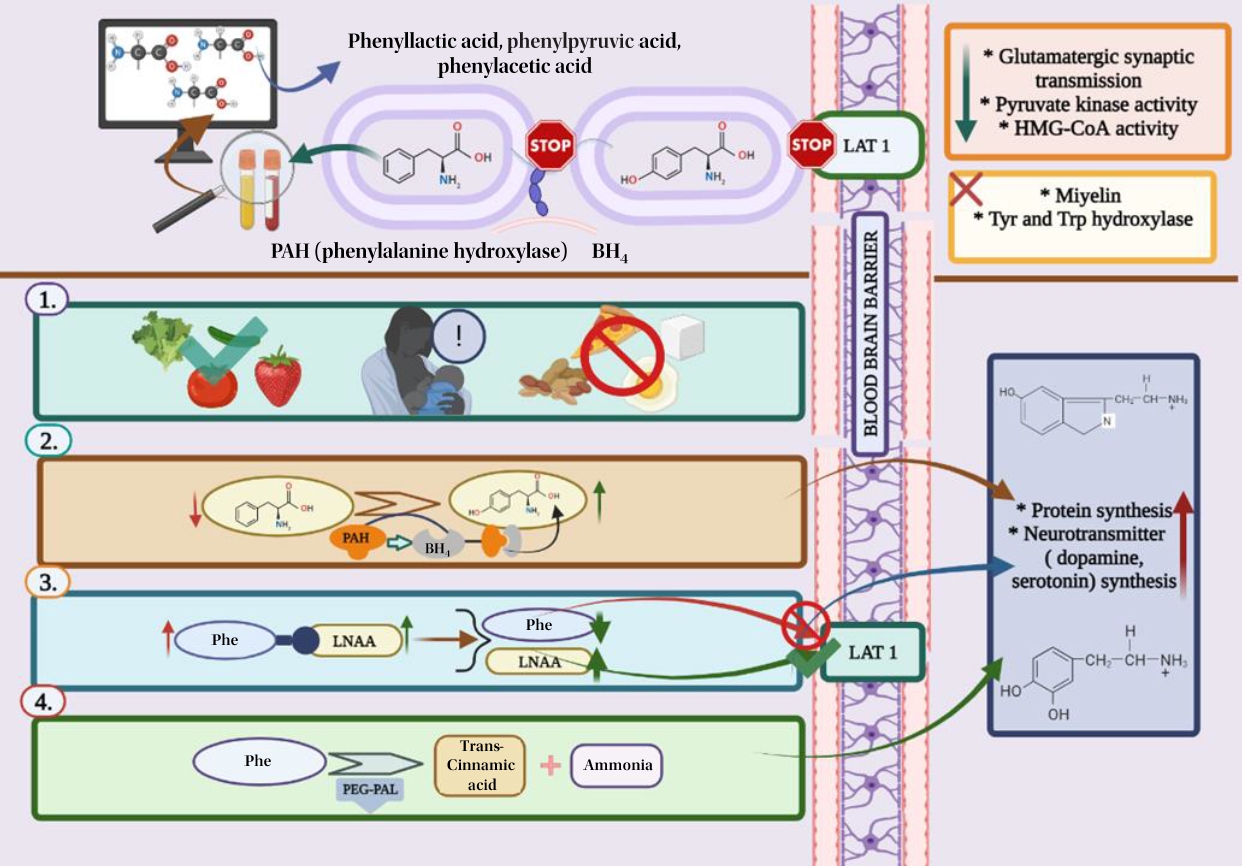
Graphical abstract. LAT1, L-type amino acid transporter 1; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; Tyr, tyrosine; Trp, tryptophan; BH4, tetrahydrobiopterin; Phe, phenylalanine; LNAA, large neutral amino acid; PEG-PAL, PEGylated recombinant. Anabaena variabilis Phe ammonia-lyase. Created in BioRender.com.
Introduction
Phenylketonuria (PKU), a genetic metabolic disease discovered in 1934 as a result of Fölling’s work on 2 Norwegian brothers with intellectual disability, is a rare autosomal recessive inherited disorder of amino acid metabolism characterized by the accumulation of phenylalanine (Phe) caused by a hepatic L-phenylalanine-4-hydroxylase (PAH) enzyme activity deficiency [1-3]. The parents of individuals with PKU have 2 genes, one intact and one defective, that are responsible for producing the PAH enzyme. Children who inherit defective genes from both parents have a 25% risk of developing PKU [4]. The approximate ratio of PKU in Caucasians is 1:10,000, and early diagnosis prevents serious and irreversible neurological sequelae [1,5]. The highest global rates belong to Europe and certain Middle Eastern nations. For example, Italy (1:4,000) and Ireland (1: 4,545) have a higher prevalence than Iran, Jordan (1:5,000), and Turkey (1:6.667). This prevalence can be explained by the higher rate of consanguineous marriages [3]. At this point, newborns are tested for PKU with the Guthrie test within the first 24–48 hours after birth, that is, after the first feeding, and if blood Phe levels exceed 6 mg/dL (360 mmol/L), the disease is diagnosed and treated [6]. Metin girmek için buraya tıklayın veya dokunun.
PKU is primarily treated with medical nutritional therapy. In this approach, natural protein sources are limited in the diet based on individual Phe tolerance, and daily protein requirements are supported by amino acid mixtures that do not contain Phe [7]. These patients do not consume primary animal protein sources, such as meat, milk, dairy products, eggs, or vegetable sources (such as oilseeds and legumes), that is, foods with high Phe content. As a natural protein source, foods containing low amounts of protein, such as vegetables and fruits, are preferred, and because of the decrease in blood Phe that results from the diet, while the use of cereals is allowed provided that they are used in limited quantities [8]. Metin girmek için buraya tıklayın veya dokunun. Simple carbohydrates and fats are important sources of energy in Phe-restricted diets [9,10]. To ensure patient growth and development, daily protein is provided using Phefree amino acid mixtures containing essential amino acids in appropriate proportions [10]. The observation of this restrictive diet is difficult for both patients and their parents and requires much planning and coordination. A number of PKU centers use casein glycomacropeptide (CGMP) or large neutral amino acids (LNAAs) as alternative dietary supplements, and some patients respond to tetrahydrobiopterin (BH4), which functions as a pharmaceutical chaperone (prescribed as sapropterin dihydrochloride) [11]. Moreover, pegvaliase (PEGylated recombinant Anabaena variabilis Phe ammonia-lyase [PEG-PAL]), which operates apart from PAH, has provided some encouraging results in adult PKU patients. It was included in different new approaches to dietary therapy by the U.S. Food and Drug Administration (FDA) in May 2018 [12].
This article discusses the primary/basic nutritional approaches implemented to treat PKU as well as the latest emerging protein substitutes, their possible efficacy, and associated safety issues.
Phenylketonuria
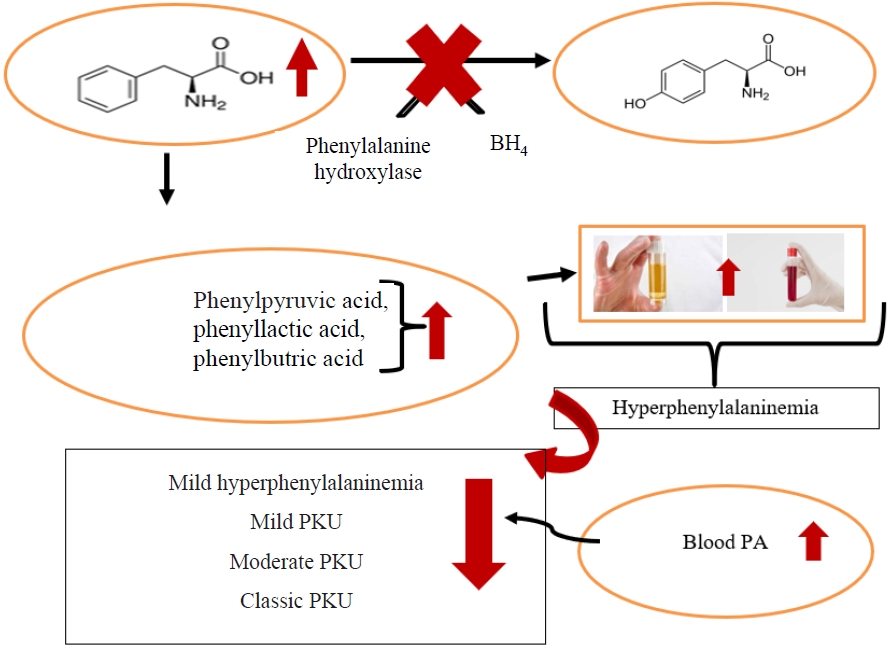
PKU is a genetically inherited condition in which amino acid metabolism is characterized by hyperphenylalaninemia, which results from a deficiency in the hepatic Phe hydroxylase enzyme. This prevents the hydroxylation of Phe, an essential amino acid, to tyrosine (Tyr), resulting in increased blood and brain Phe concentrations [13-15]. Immediate and continuous treatment after newborn screening is critical for promoting normal development and healthy growth [15]. Parents of individuals with PKU have 2 genes, one intact and one defective, that are responsible for PAH enzyme production, and children who inherit the defective genes from both parents are born with PKU, with a very high incidence of developing the disease (25%) [4,16]. The main goal of healthy growth and development is to ensure that blood Phe levels remain at a safe level and sufficient dietary intake of macro- and micronutrients [15].
Phe, an essential amino acid that is consumed as food and cannot be synthesized in the body, is converted to Tyr by the PAH enzyme in healthy individuals [16]. However, due to the absence of PAH in these patients, normal dietary Phe is not converted to Tyr, preventing the formation of important metabolites such as adrenaline, noradrenaline, thyroxine, melanin, L-dopa, and dopamine [17]. The most clinically significant pathological manifestation of PAH deficit occurs in the brain [18]. An increased blood Phe level prevents the delivery of other LNAA to the brain, which lowers their concentrations; lower LNAA concentrations may contribute to monoamine neurotransmitter deficiency by interfering with cerebral protein synthesis [19]. In the brain, Phe prevents the normal functioning of Tyr hydroxylase and tryptophan (Trp) hydroxylase, resulting in dopamine and serotonin inadequacies. Hyperphenylalaninemia is associated with anxiety and sudden mood changes.
Additionally, PAH enzyme absence or deficiency may result in the inability to metabolize Phe, an essential amino acid, and increase the levels of phenylpyruvic acid, phenylacetic acid, and phenyllactic acid, which are formed as a result of Phe and transamination in the blood and brain tissue (Fig. 1) [17,20,21]. Therefore, a remarkable feature is the musty odor caused by increased levels of phenylpyruvic acid, phenylacetic acid, and phenyllactic acid in the body fluids and urine of these patients [17,22].
When PKU is not treated early, it can cause behavioral problems, psychiatric symptoms, motor disorders, seizures, an eczematous rash, and irreversible intellectual disability [1,23,24]. Links have also been established between high Phe levels and epigenetic alterations in gene expression patterns in the brain, disrupted myelin production, impaired cerebral glucose metabolism (as seen on positron emission tomography imaging), the accumulation of amyloid plaque-like fibrils, and increased oxidative stress [13,19]. The primary nonneurological symptom of PAH deficiency is hypopigmentation (due to reduced melanin formation); in fact, light-colored hair, eyes, and skin are observed in 60% of cases [13,22].
Molecular genetics and classification
More than 1,000 mutations result in PKU, the most common of which is the substitution of arginine (Arg) and Trp at position 408 (Arg408Trp)[25]. PAH mutations that cause PKU are usually due to decreased PAH activity or expression, which causes Phe levels to increase and Tyr levels to decrease [16].
When the Arg408Trp mutation occurs, blood Phe levels exceed 1,200 µmol/L, resulting in “classic” PKU. Blood Phe values of 120–1,200 µmol/L are considered less severe mutations [16]. Classic PKU (pretreatment blood Phe, >1,200 µmol/L), mild PKU (pretreatment blood Phe 600–1,200 µmol/L), and mild hyperphenylalaninemia (pretreatment blood Phe, 120–600 µmol/L) are the 3 metabolic phenotypic groupings. The mild PKU group also includes patients with severe PKU (pretreatment blood Phe, 900–1,200 µmol/L) (Table 1) [15].
Pathophysiology
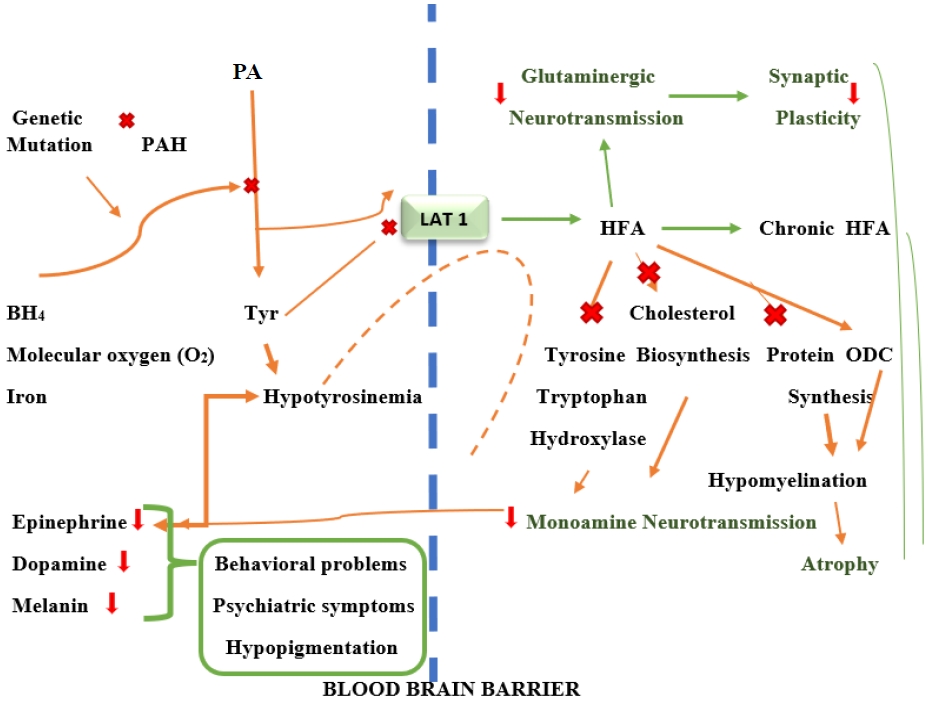
PAH is a tetrameric monooxygenase enzyme that contains iron and catalyzes the hydroxylation of Phe to Tyr (Fig. 2). This reaction requires molecular oxygen as a cofactor and reduces BH4 as a cosubstrate [25]. PAH-mediated hydroxylation is the rate-limiting phase of Phe metabolism. During regular protein cycling, 10%–20% of the standard dietary Phe intake is employed, with the remaining Phe being converted to Tyr in the presence of PAH [13,20].
PAH is responsible for producing the neurotransmitters Tyr, adrenaline, norepinephrine, and dopamine, converting thyroxine and melanin in the thyroid gland, and forming acetoacetate (a ketone) and fumarate in melanocytes (Krebs cycle intermediate) [13]. Hyperphenylalaninemia and hypotyrosinemia are diseases caused by low PAH activity. A high Phe content is associated with Trp, Tyr, and other LNAA for space on the L-type amino acid transporter 1 (LAT1) at the blood-brain barrier (BBB) and decreases serotonin and dopamine levels [16,19,20]. Furthermore, the absence or insufficiency of PAH enzymes secreted by the liver makes affected patients unable to metabolize Phe, an essential amino acid. It also leads to increased phenylpyruvic acid, phenylacetic acid, and phenyllactic acid levels as a result of Phe and transamination in the blood and brain tissue [13,17,21,26]. High Phe levels also prevent Tyr from being transported through the BBB and disrupt the Tyr and Trp hydroxylase systems as well as cholesterol synthesis, forming a hypomonoaminergic state in the limbic and frontal regions of the brain that are dominated by neurotransmitters, including serotonin and dopamine. Moreover, decreased glutaminergic neurotransmission occurs, leading to reduced synaptic plasticity and atrophy (Fig. 2) [16,19].
Treatment methods
A genetic disease involving issues with metabolization, PKU is characterized by a decreased or absent PAH activity, which causes heightened and neurotoxic Phe levels [27,28]. PAH is the first rate-limiting step in the conversion of Phe to Tyr. PKU develops from mutations in the PAH gene that produce marked hyperphenylalaninemia, brain Phe buildup, and critical damage to the central nervous system, reducing the function of PAH enzymes in untreated patients [27,29]. The primary objective for treating PKU is to safeguard Phe levels to avoid mental impairment and ensure normal growth and a healthy life until adulthood [30].
A restrictive Phe diet, the most common PKU treatment method, involves limiting natural protein sources and supplementing with low-Phe or Phe-free substitutes and specialized low-protein meals (SLPFs). The availability of certain PKU therapeutic foods varies widely among nations, and it is well recognized that adhering to a Phe-restricted diet is challenging [15,30,31]. It might be especially difficult for school-age children because it involves preparation and organization [6,30].
A variety of PKU centers utilize CGMP or LNAA as alternative dietary supplements, and some patients respond to BH4, which works as a pharmaceutical chaperone [3,11]. Individuals with milder PKU generally benefit from BH4, a pharmacological treatment. In individuals who react to BH4, it can boost natural protein intake by 2–4 times and/or lower blood Phe levels by enhancing the residual activity of the enzyme Phe hydroxylase. PEG-PAL, the most novel therapeutic option, is an enzyme replacement that converts Phe to transcinnamic acid and ammonia. It helps increase Phe tolerance by lowering Phe levels when administered as a subcutaneous injection. On the other hand, PEG-PAL is a pharmacological therapy that has been linked to immunological responses and requires careful monitoring [12,15]. Some of the old and new approaches to treating PKU are summarized in Table 2.
1. Old approaches to treating PKU
1) Medical nutrition therapy
PKU consists of an inborn error in Phe metabolism originating from a mutation in the Phe hydroxylase gene, which codes for the enzyme L-Phe hydroxylase (EC 1.14.16.1) [15]. Worldwide PKU guidelines include recommendations for target blood Phe levels that can be used to guide and monitor therapy, assess patient outcomes, and compare treatment efficacies [15]. International recommendations from various countries have established different therapeutic targets for blood Phe levels. For example, the European PKU Guidelines (2017) provided scientific data supporting a 600 µmol/L upper blood Phe target level for patients younger than 12 years. On the other hand, the US 2014 recommendations for all age categories proposed 360 µmol/L as the target for blood Phe levels [32,33].
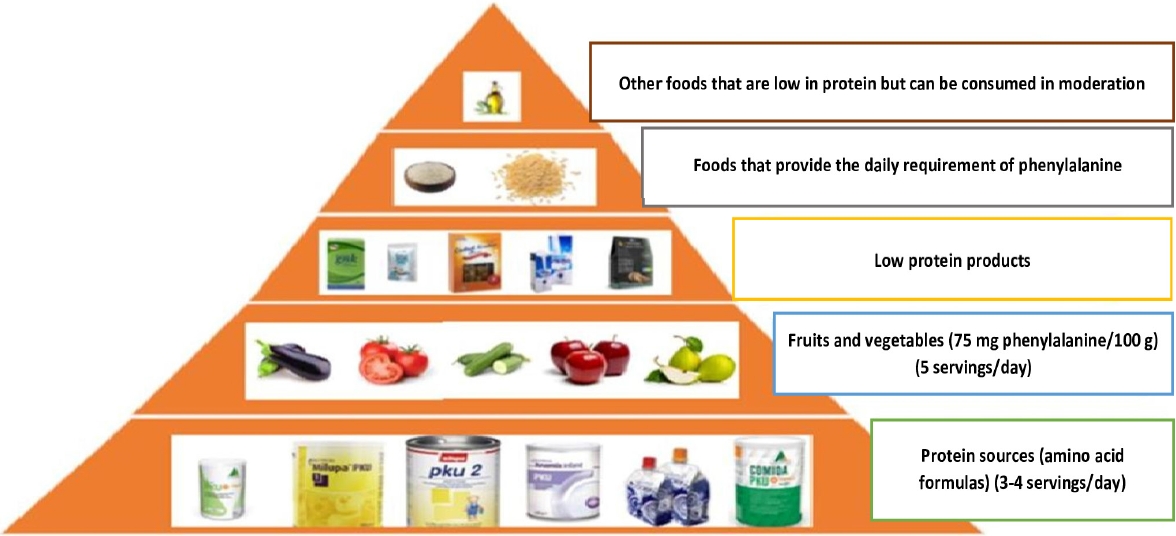
Patients with PKU are unable to process Phe because of enzymatic blockage. Consequently, a reduced Phe diet should be prescribed [15,33]. The main method of treating PKU is through medical nutrition therapy, which requires Phe in the diet to be limited according to individual tolerance and supplementation with Phe-free medicinal foods to fulfill the body’s protein requirements [6,30]. Individuals with blood Phe levels beyond particular Phe objectives should be treated with lifelong medical nutrition therapy according to established PKU management recommendations [27]. The treatment plan should be established by evaluation of a country’s specific criteria; if none exist, European or USA consensus guidelines can be applied [5,6]. Three phases of dietary therapy, namely, a low-protein diet, Phe-free therapeutic foods, and genetically modified foods with a low-protein constitution, are included in these instructions (Fig. 3) [6].
While under treatment, patients with PKU should avoid protein-rich foods (dairy products, meat, and fish, for example) and primarily consume natural foods with a low-protein content, such as fruits and vegetables, and products such as low-protein breads and pastas. In addition, their protein intake should be adjusted individually according to age, body weight, and Phe tolerance to promote optimal growth and development without increasing blood Phe levels [5,6,27].
Phe-free amino acid formulations are used in medicinal foods to add alternative proteins and minerals. Formula supplements are necessary to maintain an adequate protein intake [6,27]. There are a wide array of formulas on the market that include both gel and ready-to-drink options [6]. In addition, the required daily protein is obtained from protein substitutes that do not contain Phe but do contain essential amino acids in appropriate proportions [5,6,27]. These foods help provide the energy, protein, fat, and other essential nutrients needed to maintain optimal health status.
A low-protein diet is one treatment option for PKU. Protein levels decrease in modified low-protein diets [2]. Specific items can be used to minimize protein consumption. Low-protein meals are available in various forms and combinations that can help people obtain enough energy and adjust their diet appropriately [6]. A 3-year study evaluating SLPFs in individuals with PKU reported that low-protein milk, pastas, and breads supplied the most energy, while the intake of sweet SLPFs (chocolate, cakes, biscuits, etc.) was low [34]. Although improvements in taste, presentation, suitability, and nutritional value of dietary substitutes have improved long-term adherence to the PKU diet, the suitability of these alternatives is not fully confirmed and greater research is needed in this field [27].
Low-protein products are quite expensive, which can decrease their accessibility to families. In addition, insurance companies and government agencies do not always cover the necessary medicinal foods, which may lead to a significant financial burden on families [6]. Furthermore, in neonates with Phe levels greater than 600 µmol/L, diet therapy should begin soon after the PKU diagnosis is confirmed and should be continued for life. Infancy and childhood treatment compliance are sufficient, but challenges maintaining a PKU diet during adolescence and adulthood have been described [6,11,27].
When incompatibility is combined with higher circulating Phe concentrations and consequently poorer metabolic management, patients may be at risk of nutritional inadequacies compared to well-adjusted individuals and healthy controls [6]. Despite compensatory increases in dietary protein intake, patients who discontinue PKU diets have insufficient nutritional intake [27]. Inadequate intakes of Fe, Ca, Zn, I, Se, vitamin B12, and long-chain polyunsaturated fatty acids (especially docosahexaenoic acid and arachidonic acid) has been reported. Disturbances in cognitive and behavioral functions (attention and reaction time) and mood (low self-esteem, depression, anxiety, and increased inhibition) also occur as a result of the pathophysiological effects of impaired Phe hydroxylation in PKU patients who do not comply with treatment [28,35].
2) Phe tolerance
Phe tolerance is defined as the amount of Phe consumed by a person with PKU that maintains therapeutic blood Phe concentrations [6]. Blood Phe levels should be 120–360 µmol/L for children under the age of 12 years and women following the diet before or during pregnancy and 120–600 µmol/L for patients older than 12 years [9].
Dietary protein tolerance is often less than 10 g/day (500 mg/day) in individuals with PKU. To achieve this, dairy products, meat, and high-protein plant-based meals must be avoided [2]. Both patients and physicians must use substantial problem-solving techniques to promote PKU dietary adherence [10].
Tolerable dietary Phe amounts vary according to disease severity (those with mild or moderate PKU are able to process more protein), dosage of protein substitute, adherence, daily distribution, and medication therapy, including sapropterin or PEG-PAL (for patients >16 years of age) [11]. Growth is also affected by a catabolic state during pregnancy and disease. Patients responding to sapropterin are expected to double their Phe tolerance to at least 500 mg/day, the safe and reasonable protein intake level defined by the World Health Organization/Food and Agricultural Organization of the United Nations/United Nations University [35].
New approaches to the treatment of PKU
Patients with PKU must maintain a strict diet for life. Treatment aims to lower blood Phe levels to reduce the neurocognitive and mental consequences of PKU [9]. Nutritionists, psychologists, social workers, and metabolic experts should collaborate to offer multidisciplinary care to individuals with hyperphenylalaninemia and PKU [2,6]. Following a restrictive diet supplemented with Phe-free medicinal foods is the most noteworthy method for preventing nutritional deficits [6,23]. Therapeutic foods are Phe-free and Tyr-enhanced protein substitutes that may contain CGMP and other major neutral amino acids as protein sources [30]. Following such a stringent diet will undoubtedly be difficult for patients and entail special planning and coordination. At this point, diet has become a central focus for those with PKU and their caregivers [6,9]. In addition, parents state that they have problems adapting their children to their diet starting at school age [6,30]. The current study aimed to discuss the outcomes of new approaches used in the treatment of PKU and determine whether they are an alternative to diet therapy, thereby creating a basis for future studies. Each newgeneration protein substitution is discussed under separate headings in this context.
1. BH4 treatment
BH4 functions as a critical cofactor in the PAH enzyme system by supporting correct folding and stability of the PAH enzyme [19]. Although the FDA approved sapropterin dihydrochloride for the treatment of hyperphenylalaninemia in patients with BH4-responsive PKU or BH4 deficiencies in 2007 and the European Medicines Agency followed its lead in 2008, significant inter- and intragroup differences have been noted due to the wide variety of protocols used in clinics, especially the BH4 response test (Table 3) [2,32,36-38]. It was later authorized for use in all patients, including children under 4 years of age, by both regulatory bodies [23]. At this point, sapropterin dihydrochloride is the first pharmacological treatment for both pediatric and adult patients with PKU who respond to BH4 in conjunction with dietary therapy. However, as with any pharmacological treatment, caution is recommended regarding its use during pregnancy [34].
According to the American College of Medical Genetics and Genomics, sapropterin is typically administered once daily at 5–20 mg/kg, although the preferred dosage is 20 mg/kg. Although testing is usually performed at the time of the first diagnosis in Europe, this application is not active in the US. If the test is performed during infancy in the US, it is advised to first reduce blood Phe levels to 480–600 µmol/L. A sapropterin response is usually established after noting a baseline blood Phe level on the same day of the drug initiation (baseline) and then starting a daily course of 20-mg/kg sapropterin to ensure that the patient does not underestimate the response rate [15,32]. It is then evaluated using regular blood Phe levels at regular intervals, usually at 24 hours, 1 week, 2 weeks, and even up to 3 or 4 weeks in some instances. When treatment is applied alongside constant dieting during the test period, the patient appears to respond to treatment when a significant drop in blood Phe is noted. Most patients responding to sapropterin experience a rapid reduction in blood Phe levels, although some patients experience a delay of 2–4 weeks. For patients able to maintain a reasonable blood Phe level through only diet therapy, the most significant benefit of sapropterin therapy is the greater amount of natural protein in the diet by increasing Phe tolerance and dietary protein in more susceptible patients [32].
Without a consensus-defined schedule for BH4 challenge testing, it funs 24–48 hours to several administered BH4 doses (10–20 mg/kg) every 24 hours in Europe [30,34]. In Italy, a 48-hour schedule for BH4 challenge for newborn screening has been widely adopted as 10 or 20 mg/kg of sapropterin, although protocols for testing BH4 differ slightly among centers. The European Group reached a consensus, whereby a traditional 24-hour protocol for challenge testing with 20-mg/kg sapropterin was implemented for differential diagnosis in newborns [11,39,40].
Most current European guidelines suggest that genotype and metabolic phenotype are highly indicative of BH4 response; therefore, all patients should be evaluated for BH4 response using a BH4 response test or genotyping [39,40]. Patients with high residual activity of the PAH enzyme, as well as those with BH4 deficiency, are more likely to respond to BH4, but few patients with classic PKU benefit from BH4 therapy [41,42]. Patients suitable for BH4 treatment can be identified using the BH4 response test [30,35]. Not all patients respond to sapropterin treatment; therefore, they must be tested [23]. This suggests that numerous Phe levels should be determined before and after the start of BH4 therapy, and a 30% or greater decrease in blood Phe levels from baseline indicates a treatment response [3,19]. Those with adequate metabolic management respond to BH4; sapropterin dihydrochloride therapy significantly lowers blood Phe concentrations, increases natural protein intake, and increases patient dietary management and compliance [34].
Evidence from both systematic reviews also shows that BH4 effectively reduces blood Phe concentrations, increases Phe tolerance among PKU patients responsive to sapropterin dihydrochloride, and appears to help improve the diet therapy of patients [19,31]. For example, the SPARK extension study found that, in children under 4 years of age, long-term sapropterin treatment alongside a Phe-restricted diet maintained a boosted dietary Phe tolerance that can last for more than 3.5 years [43].
A randomized-controlled (RCT) trial evaluating BH4 therapy reported that it did not appear to affect Phe variation in a small sample of BH4-responsive PKU patients. In this study, the variation in daily Tyr and Phe/Tyr ratio was lower during BH4 treatment. This effect may be due to an increasing trend in fasting Tyr levels or BH4 treatment. Therefore, BH4 treatment followed by dietary liberalization demonstrated that Phe concentrations affect Tyr synthesis and variability and may therefore be an effective PKU treatment by improving Phe/Tyr ratios [44].
In 2 RCT, BH4 treatment dramatically lowered Phe levels without major side effects compared with placebo [45,46]. Patients who responded successfully experienced enhanced Phe tolerance, which allowed them to replace low-protein foods with 2–3 meals of bread and cereals [45]. However, only a small percentage of those with PKU can eat whatever they want, and the use of low-Phe therapeutic products tends to be necessary to meet their nutritional needs. A retrospective analysis of 147 patients with mild or classic PKU treated with BH4 for more than a decade revealed improved dietary adherence in 47% and improved treatment adherence in 63% [47]. Overall, it appears to be a moderately favorable short-term effect of BH4 on decreasing Phe levels and enhancing metabolic control in various PKU groups; the possible long-term impacts of BH4 on cognition and/or quality of life remain unknown, and research continues [31].
2. Casein glycomacropeptide
CGMP, a polar glycophosphopeptide, is composed of 64 amino acids, is free of aromatic amino acids (Phe, Trp, and Tyr), and contains more substantial concentrations of threonine and isoleucine than other dietary proteins [11]. CGMP, an incomplete protein manufactured naturally during the cheese-making process, is commercially used as a nutritional ingredient for various applications. When used to treat PKU, it is supplemented with essential amino acids and Tyr to create a substitute for normal daily protein, and the availability of highly pure CGMP containing no more than 2 mg/g of Phe is necessary before CGMP medicinal foods can be formulated [11,27]. Commercial protein substitutes comprise 60%–70% of the protein equivalent from CGMP and the remaining protein source from traditional amino acids [11].
Sensory studies of patients with PKU demonstrated that CGMP medicinal foods improve protein retention, nitrogen utilization, neurophysiology, BBB Phe control, satiety, and bone strength, demonstrate immunological activity owing to their prebiotic properties, and contribute to food taste and acceptance among patients with PKU [30,48,49]. CGMP comprises 20% –25% of the total protein content of whey products. Additionally, research has shown that whey protein provides more satiety than other sources of protein such as casein, soy, and egg albumin. CGMP influences the hormone responses that affect satiety [50].
In one 3-year longitudinal study evaluating the efficacy of a CGMP-amino acid (CGMP-AA) supplement among children with PKU, CGMP-AA did not seem to affect energy intake, body weight gain, or body mass index but indirectly affected satiety compared to AA supplementation alone. The effect of CGMP-AA on satiety, especially when combined with other foods related to the amount and timing as well as satiety signals, has not been fully established and requires further study [51]. Moreover, in a longitudinal study spanning 3 years in pediatric patients with PKU, no substantial change in body composition was noted between the CGMP-AA and AA groups, whereas evidence of body composition improvements in the group receiving CGMP-AA was noted [52].
To evaluate the sensory properties of products containing low-Phe amounts, 8 low-protein recipes, each containing a protein equivalent of 5 g/100 mL, were used, such as the CGMP formula (nutrient composition: 9 g of carbohydrates, including 7 g of sugar; 1.4 g of fat, and 5 g of protein) and the L-amino acid formula (nutritional composition: 43 g of carbohydrates, including 3 g of sugar; 14 g of fat, and 5 g of protein per 100 g), in 86 patients with PKU (45 men, 41 women) on a Phe-restricted diet at a hospital in Italy. The acceptability of CGMP beverages as the principal source of protein in the PKU diet was greater than the currently available AA formulas especially in young people [27]. CGMP also supports and positively impacts its ability to improve the overall dietary variety, taste, glucose reponse, compatibility and widen the array of palatable foods and beverages [53,54]. In terms of diversity, one of the most significant barriers to the observation of PKU diet is that amino acid blends tend to be consumed in liquid form [55]. CGMP can be ingested via semi-solid food products and beverages [56]. In fact, a wide variety of low-Phe foods, such as puddings, beverages, and crackers, have been used for some time as suitable foods owing to their functional properties, including good heat stability and acid solubility [27].
Studies in a murine mouse model of PKU (Pah enu2) showed that CGMP provides an ample amount of protein that supports lean body mass, decreases body fat mass, and markedly increases fat oxidation when paired with AA supplementation. Research on murine mice and humans with PKU suggests that CGMP is a physiological source of low-Phe dietary protein as opposed to synthetic AA [48,49].
In a study of 11 patients aged 11–31 years, the effect of Phe-free 1-AA and CGMP-AA on PKU was evaluated using 4-day treatment [53]. Although no major intergroup difference was evident in postprandial Phe concentrations, fasting Phe concentrations were considerably lower with CGMP-AA than with l-AA. In conclusion, it was quite clear that CGMP-AA is a reliable alternative to 1-AA-based protein substitutes [53]. Furthermore, when insulin and appetitive hormone ghrelin plasma concentrations were measured before and 180 min after breakfast in the same patient group, concentrations were significantly lower after CGMP-AA supplemental, which created a greater feeling of satiety [57]. CGMP stimulates the release of cholecystokinin, a peptide that can promote satiety. However, further research in pediatric populations is required to assess whether this protein source is safe for children [11].
3. Large neutral amino acids
LNAA was initially proposed as a treatment option for patients with PKU in 1948 [11]. LNAA are protein substitutes administered to adult patients who do not fare well under a low-Phe diet [58]. LNAA is considered a new protein substitute because the transport of Phe between the intestinal mucosa and BBB can reduce Phe concentrations through competitive inhibition by LNAA type 1, which has a high affinity for Phe expressed in the brain [31,48]. All LNAA carried by this transporter contain branched-chain amino acids (valine, leucine, and isoleucine), aromatic amino acids (Phe, Tyr, and Trp), and other amino acids, including histidine, threonine, and methionine, which are indispensable for protein synthesis in PKU [31]. There is limited evidence to confirm the ability of LNAA supplementation to reduce blood Phe levels and improve cognitive outcomes [41]. Owing to insufficient evidence regarding the long-term effects of LNAA therapy, it is now considered a short-term therapy option. However, dietary therapy forms the basis of PKU management, upon which new treatments have been developed [59].
An RCT showed a significant decrease in blood Phe concentrations among patients with PKU who had been treated with LNAA for 2 weeks [60]. This study recommends including LNAA in PKU diets to reduce blood Phe concentrations [60]. Furthermore, a study of PKU mice reported reduced brain LNAA concentrations and concluded that restoring large neutral amino acid levels in the brain may improve cognitive outcomes [61]. Similarly, in another study, 45 female mice testing homozygous for the Pahenu2 mutation were treated over 12 weeks in various groups of different CGMP and LNAA combinations, and it was reported that mice treated with CGMP or combination CGMP and LNAA had significantly lower brain and plasma Phe concentrations compared to mice who received casein only. However, extra LNAA in CGMP had no critical impact on plasma or brain Phe levels, whereas it increased brain serotonin levels [62].
To explain the relationship between plasma Phe levels and brain biochemistry, blood AA, brain Phe, and monoaminergic neurotransmitter levels were evaluated. The researchers demonstrated a substantial correlation between plasma Phe concentrations and brain Phe concentrations; however, these values were considerably inversely correlated with brain serotonin and norepinephrine and very weakly correlated with brain dopamine. This suggests that dopamine may be less affected in PKU patients or mice than currently believed [63]. Another study was conducted to determine the added/protective effect of LNAA supplementation to a less severe versus severe Phe-restricted diet on plasma and brain AA and neurotransmitter concentrations in adult PKU mice. This was in response to the fact that many adult patients with PKU do not meet the terms of the strict dietary restrictions. In addition, LNAA supplementation to a less severe Phe-restricted diet improved brain neurotransmitter and Phe concentrations. Compared to a strict Phe-restricted diet, it was equally effective at restoring brain norepinephrine and serotonin concentrations but less effective at reducing brain Phe concentrations [64].
The ability of LNAA supplementation to lower blood Phe levels remains unknown, and studies to date featured short study periods and small patient cohorts using varying dosages (250–1,000 mg LNAA/kg/day) and different LNAA formulations [65].
A new slow-release formulation rich in Tyr and Trp was tested as an LNAA product in a study conducted over a 12- month period. The patients’ blood Phe levels did not change during the study. Moreover, the study found reductions in Phe levels even over durations shorter than 6 months, but most returned to baseline at the end of the 12-month treatment period. Studies have indicated that this is a viable treatment for adult patients [66].
LNAA (but low threonine) supplementation in PKU mice dramatically reduced brain Phe concentrations and neurological issues compared to low LNAA-contained control diet, although not in all mice. However, in PKU mice, LNAA supplementation increased brain serotonin and norepinephrine concentrations in wild-type mice from 35% to 71% and from 57% to 86%, respectively, but did not increase brain dopamine concentrations [64]. Furthermore, in another study using ten 6-month-old PKU mice as a model for adult PKU patients, the substitution of a synthetic Phe-free amino acid mixture for LNAA supplementation in PKU mice on a less severe Phe-restricted diet significantly increased brain norepinephrine and serotonin levels. Moreover, reduced Phe concentrations reportedly led to healing in the brain [67].
Nevertheless, the ideal composition of LNAA has yet to be determined. It is possible that patients receiving Phe-free 1-AA dietary supplementation may experience no additional benefits [31,68]. However, the current evidence is certainly not infallible, and additional research is needed to demonstrate that LNAA is a viable approach to lowering Phe levels or increasing dietary Phe tolerance without adversely affecting kidney and bone health [31]. LNAA supplementation is mainly considered among elderly patients who cannot maintain dietary compliance [69]. It is not routinely available in all centers in Europe and has not been tested in children under 11 years of age. As no safety data are available, it should not be administered during pregnancy [11].
Briefly, LNAA supplementation alone or in conjunction with a low-Phe diet is able to improve the health outcomes of individuals who struggle to meet the demands of a strict low-Phe diet. However, there is an ongoing need for more long-term studies evaluating the efficacy and safety of LNAA supplementation [28].
4. Pegvaliase
PEG-PAL was recently developed as an enzyme substitution therapy to reduce blood Phe concentration in adults with PKU [11,34,70]. PEG-PAL converts Phe to ammonia, which is metabolized by the liver, excreted in the urine, and then converted to transcinnamic acid [11,28,71]. PEG-PAL therapy is a reasonable course of action for adult PKU patients (>16 years) with blood Phe levels above 600 µmol//L on current therapy [28]. In clinical studies, PEG-PAL demonstrated high clinical efficacy at lowering and maintaining blood Phe levels within target ranges while allowing a diet with intact protein intake close to that recommended for the general population. Moreover, improvements in inattention and mood symptoms were observed with PEG-PAL therapy [11,35].
In a phase 1 trial, 25 adult patients with PKU were administered increasing doses (0.001–0.100 mg/kg) of recombinant PAL-PEG (rAvPAL-PEG), which most commonly featured the side effects of infection and dizziness. The mean blood Phe concentration decreased by 54.0% in 5 patients who were administered the highest dosage [71] In a phase 2 extension study, the administration of rAvPAL-PEG to 67 subjects with PKU for 1 year reduced blood Phe concentrations by a mean 65% compared to pretreatment levels in patients with a Phe level <600 µmol/L [72]. PEG-PAL did not significantly reduce blood Phe levels among individuals with PKU in 2 open-label multisite prospective phase 2 dose-finding investigations (PAL-002 and PAL-004). The first phase 2 trial used a low initial dose of PEG-PAL to address tolerability concerns [73].
PEG-PAL has been studied in more than 350 adult patients with PKU in 7 different clinical trials, including the PRISM trial, the largest phase 3 clinical program conducted in adult patients with PKU [70]. A phase 3, 8-week double-blind RCT was conducted with 86 patients either staying on rAvPAL-PEG or receiving a placebo. The rAvPAL-PEG-treated group maintained the lowest blood Phe level (~500 µmol/L), while that of the placebotreated group increased to 1386 µmol/L. It is estimated that patients in this study could consume 75.0% of the suggested amount of protein for a normal adult. No improvement in attentiveness or mood disorders was noted, and adverse events such as hypersensitivity, arthralgia, headache, and fatigue were more common in patients treated with rAvPAL-PEG than placebo [70,74]. Such treatment has only been used in adult patients with PKU, although it reportedly reduces Phe concentrations and cause some side effects [11].
Although PKU is one of the most well-studied inborn metabolic disorders, the current treatments often fail to reduce blood Phe levels, resulting in significant morbidity. PEG-PAL is a potential new drug that lowers blood Phe levels and addresses the metabolic etiology of PKU. With increased natural protein intake, several participants in PEG-PAL clinical trials attained blood Phe levels of 360 µmol/L. The open-label extension of the phase 3 study is ongoing and will continue to evaluate its long-term safety and efficacy [70].
Major clinical studies revealed that patients maintained their diet and dietary protein consumption (from natural or pharmaceutical sources) within 10% of that of their previous diet during PEG-PAL treatment [71,72,74]. Blood Phe levels should be measured every 1–4 weeks throughout dose and diet titration, and the risk of hypophenylalaninemia should be considered. If necessary, one may reduce the PEG-PAL dose or increase the dietary protein intake [31].
PEG-PAL determines the immunogenic response in all patients in both early (first 6 months) and late (last 6 months) treatment by producing both anti-PEG and anti-PAL antibodies, and the presence of circulating anti-drug antibodies creates drug–antibody complexes that are more significant during early PEG-PAL therapy and slowly decrease over time. Therefore, the onset of therapy and titration are essential for the patient’s immunological response, and probable hypersensitivity should be considered at these times [28,31].
5. Gene therapy
Over the last 2 decades, research on PKU treatment has begun to incorporate gene therapy [28]. Gene therapy, a possible treatment option for PKU, has been examined by several researchers. A mouse model of PKU was recently administered a recombinant adeno-associated virus (rAAV) encoding the PAH gene. The rAAV contrast, the rAAV vector, was lost during hepatocyte regeneration when it failed to integrate into hepatocyte genomes and thus failed to permanently correct hepatic PAH. The muscle could convert Phe to Tyr when the vector containing the genes producing PAH and cofactors was inserted, but the incomplete gene transfer suggests that the strategy requires improvement [25,28]. Studies of mouse models of PKU demonstrated that gene therapy can also be effectively applied to nonhepatic tissues such as muscles. The addition of vectors containing the genes required for synthesizing PAH and BH4 to muscle cells resulted in a system capable of converting Phe to Tyr, which imitates the role of hepatic Phe metabolism [25,28].
Another method is to use aminoglycosides to detect PAH nonsense mutations in vitro. Aminoglycosides, such as gentamicin and G-418, can encourage the reading of stop codons, resulting in protein synthesis. Unfortunately, none of the mouse models for PKU to date include a genetic background (variation) that allows researchers to determine the level of reading in the liver’s sufficiency in restoring Phe tolerance. Additional research will help uncover the effects of this strategy on PKU treatment [28].
Conclusion
PKU develops when the PAH gene undergoes mutations and is compatible with BH4, the primary cofactor that converts Phe to Tyr. Therefore, lifelong medical nutrition therapy is recommended for these patients. Dietary therapy is 3-staged and includes a diet low in protein, use of Phe-free medicinal foods, and inclusion of low-protein foods modified to meet the prerequisites of the condition. However, it is difficult to observe the PKU diet in real life, as such restrictions can become a hindrance. Therefore, many adolescents and adults cannot successfully comply with their dietary demands. At this point, new protein substitutes such as sapropterin, CGMP, LNAA, PEG-PAL, and gene therapy are critical for improving dietary compliance.
Although sapropterin dihydrochloride therapy, which is currently the most preferred new protein substitution approach, is highly effective at reducing blood Phe levels, the degree of evidence in support of the enormous positive effect of BH4 on lowering Phe levels and improving metabolic control in a particular group of patients in the short term is moderate, and the potential effects on long-term health outcomes such as cognition and/or quality of life have not been uncovered; therefore, relevant studies are ongoing.
Sensory studies in individuals with PKU have shown that CGMP medicinal foods effectively improve protein retention and nitrogen utilization, neurophysiology, BBB Phe control, taste, and food acceptance among patients with PKU. However, more research is needed to determine whether this source of protein substitutes is safe for children.
It is currently difficult to determine how well LNAA supplements are able to reduce blood Phe levels among PKU patients, as its effects are only monitored for short durations using varying dosages (250–1,000 mg LNAA/kg/day) and different LNAA formulations in a limited number of patients for the short term. Therefore, long-term outcome studies evaluating the efficacy and safety of LNAA supplementation are required. Treatment with PEG-PAL has been associated with statistically significant improvements in overall safety and blood Phe concentrations but causes side effects such as hypersensitivity, arthralgia, headache, and fatigue. This treatment has been performed in adult patients with PKU, is very effective at lowering blood Phe concentrations, and causes several side effects. Accordingly, the initiation of therapy is critical to the patient’s immune response and requires special attention to the potential for hypersensitivity at these stages.
In conclusion, new approaches for protein substitution are promising. However, long-term clinical studies are required to evaluate their efficacy (optimal dosage and conditions) and safety.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


