

Introdction
Glucocorticoid (GC) is an important chemotherapeutic agent used to treat leukemias and lymphomas1-3). GC combines with a cytoplasmic glucocorticoid receptor (GR) that binds to specific DNA sequences as a transcription factor. The liganded receptors also bind to and interfere with other transcription factors, such as activator protein-1 and nuclear factor-[kappa]B, and inhibit the mitogen-activated protein kinase pathways that mediate the expression of many of the genes involved in inflammatory and immune responses, including apoptosis and cell cycle arrest4, 5). Clinically, GC sensitivity is an important factor determining prognosis of acute lymphoblastic leukemia (ALL), and non-responsiveness to GC is used as a marker for classifying patients into risk groups1, 6). However, the molecular mechanisms of the anti-leukemic effects and the clinically important phenomenon of GC resistance require further study4, 5, 7).
Progression from the G1 to S phase of the cell cycle is regulated by a series of structurally related enzymes: cyclin regulates the activation of cyclin-dependent kinases (CDKs); CDKs regulate the retinoblastoma protein (pRb) and induce the subsequent release of E2F transcription factors and expression of the genes required for the S phase. Cyclin-CDK complexes are regulated negatively by a family of kinase inhibitors8, 9). The p16INK4 (CDKN2A) gene located on chromosome 9p21 encodes a protein (p16) that inhibits CDKs and that can block cell cycle progression8-10). Frequently, p16 is mutated or inactivated in primary tumors, including leukemias, lymphomas, gliomas, lung carcinomas, and many cancer cell lines; it is thus regarded as a tumor suppressor gene11-13). Recently, some studies have reported that p16 is associated with the prognosis of hematologic malignancies, although this is controversial11). It has also been suggested that p16 has a role in GC-related apoptosis in leukemic cells, and the inactivation of p16 in B-cell ALL may induce cells that are more resistant to GC14, 15). However, few studies have examined the relationship between GC responsiveness and p16, and the roles and exact mechanisms of p16 in hematologic malignancies are not clear.
This study evaluated the relationship between GC responses, including GR expression and subsequent apoptosis, and p16, using the B-cell lymphoblast cell line NC-37.
Materials and methods
1. Cell line, culture conditions, and reagents
The B-cell lymphoblast cell line NC-37 (ATCC number, CCL-214) was purchased from ATCC (Rockville, MD, USA). NC-37 cells were maintained in RPMI-1640 medium (Gibco BRL Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco BRL, Rockville, MD, USA) at 5% CO2 and 37℃ at saturated humidity.
2. p16 siRNA transfection
For p16 siRNA transfection, a commercial kit was used (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Briefly, the following solutions were prepared: solution A contained 7 µL p16 siRNA (sc-36143) or control siRNA (sc-36869 or sc-37007) in 100 µL siRNA transfection medium (sc-36868). Solution B contained 6 µL siRNA transfection reagent (sc-29528) in 100 µL siRNA transfection medium.
Solution A was added to solution B directly, and the mixture was incubated for 30 min at room temperature. For each transfection, 0.8 mL siRNA transfection medium was added to each tube containing the siRNA and transfection reagent mixtures. In 6-well tissue plates, 3×105 cells were seeded per well and the mixtures were overlaid onto the cells. The cells were incubated for 6 h at 37℃ in a CO2 incubator, and then the transfection mixtures were removed and replaced with RPMI-1640 medium supplemented with 10% FBS and then incubated for an additional 6 h.
3. Western blot analysis
Western blotting was used to detect p16 protein after p16 siRNA transfection. Experiments were done for wild-type, control, and p16 siRNA-transfected NC-37 cells. The cells were collected by centrifugation, washed in phosphate-buffered saline (PBS), and lysed by the addition of SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 6% [w/v] SDS, 30% glycerol, 125 mM DTT, and 0.03% [w/v] bromophenol blue). Total cell samples were lysed and denatured by boiling for 5 min at 100℃. Equal amounts of protein from each sample were separated by 15% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked for 1 h with Tris-buffered saline containing 5% (w/v) milk and 0.1% Tween 20, then incubated with the primary rabbit monoclonal antibody for p16 (p16 INK4A Antibody; Cell Signaling Technology, Beverly, MA, USA) overnight at 4℃. The blots were washed with Tris-buffered saline containing Tween 20, incubated with the anti-rabbit secondary antibody (Cell Signaling Technology, Beverly, MA, USA) for 2 h, and developed using West-Zol TM plus (iNtRON Biotechnology, Seoul, Korea).
4. Cell culture with dexamethasone (DX)
DX purchased from Sigma Chemical (St. Louis, MO, USA) was dissolved in dimethyl sulfoxide and added to the medium after p16 siRNA transfection. To study the effect of DX on p16 status, we added DX to samples containing wild-type, control, and p16 siRNA-transfected NC-37 cells. The final concentration of DX was adjusted to 100 nM. For measurements, cells were harvested 6, 12, 18, and 24 h after DX addition and then prepared for the next steps.
5. Flow cytometry analysis of GR expression
Cultured cells were washed twice in PBS containing 1% bovine serum albumin. Aliquots of 1×106 cells were fixed in paraformaldehyde at room temperature for 30 min, washed, and permeabil ized with 0.1% Triton X-100 in 0.1% citrate buffer for 5 min on ice. The cells were washed twice and incubated at 4℃ for 70 min with either FITC-conjugated anti-GR antibody (5E4; AbD Serotec, Oxford, UK) or FITC-conjugated isotype control (IgG1 AbD Serotec, Oxford, UK). Finally, the cells were washed and resuspended in PBS containing 1% bovine serum albumin and analyzed using a Coulter Elite flow cytometer (Beckman Coulter Inc., Fullerton, CA, USA).
6. Cell apoptosis test
To detect apoptosis, we performed double staining with FITC-Annexin V and propidium iodide (PI; R & D Systems, Minneapolis, MN, USA). First 1×106 cells were washed with PBS. FITC-Annexin V was diluted at a concentration of 1 mg/mL in binding buffer, and the cells were resuspended in 1 mL of this solution (prepared fresh each time). The resuspended cells were incubated for 10 min in the dark at room temperature, and then 0.1 mL PI solution was added to the cell suspensions before analysis to give a final concentration of 1 mg/mL. These cells were analyzed on using a Coulter Elite flow cytometer. Annexin V single-positive cells were regarded as early apoptotic cells, whereas Annexin V/PI double-positive cells were regarded as late apoptotic cells.
7. Alamar blue (AB) assay for cell viability
First 1×104 cells were suspended in 95 µL phenol red-free RPMI-1640 containing 0.1% FBS, then seeded into 96-well plates. DX was added to each plate at a concentration of 100 nM. After 6, 12, 18, and 24 h, 11 µL AB solution was added to the medium directly, resulting in a final concentration of 10%. As a negative control, AB was added to medium without cells. Then the plates were incubated for 4 h at 37℃. The absorbance of test and control wells was read at 570 and 595 nm with a standard spectrophotometer. The number of viable cells correlated with the magnitude of dye reduction and is expressed as a percentage of the AB reduction compared to control AB.
8. Statistics
All statistical analyses were conducted using SPSS 13.0 (SPSS, Chicago, IL, USA). The results were expressed as the mean±standard deviation, and the differences between the cell groups at each time point were analyzed using the Mann-Whitney U-test. General repeated measured analysis of variance was applied to identify the trends of time-dependent GR expression, apoptosis, and cell viability between the two groups. P value <0.05 was regarded as statistically significant.
Results
1. Identification of p16 RNA interference in NC-37
To determine whether p16 affects GR regulation and the apoptosis of lymphoblast cells, we generated p16 siRNA-transfected NC-37 cells that did not express p16. First we used the florescence-expressing control siRNA to confirm p16 siRNA transfection. When more than 50% of the control siRNA-transfected NC-37 cells had fluoresced, Western blot analysis was performed to detect p16. The wild-type and control NC-37 cells expressed p16 in the immunoblot analysis, whereas the p16 siRNA-transfected NC-37 did not (P<0.05, Fig. 1).
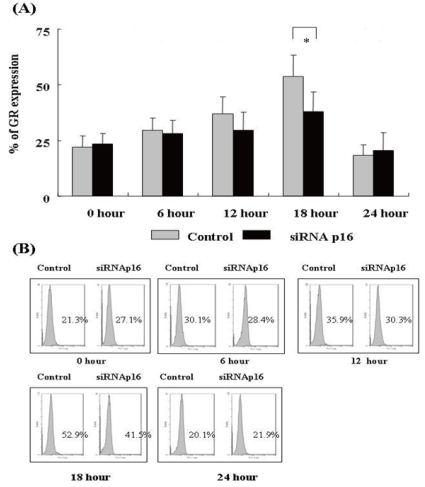
2. GR expression after DX treatment
We evaluated time-dependent GR expression 0, 6, 12, 18, and 24 h after treatment with DX for control (p16+ NC-37) and p16 siRNA-transfected (p16- NC-37) cells. The GR levels were determined by flow cytometer after intracellular GR staining at each time point. After DX treatment, GR expression began to increase after 6 h, reached a peak at 18 h, and decreased sharply by 24 h (P<0.05). The GR expression levels at 18 and 24 h in both groups differed statistically (P<0.05). The degree of GR expression tended to be higher for p16+ NC-37 cells than p16- NC-37 cells at all times, and the difference was significant at 18 h (P<0.05, Fig. 2).
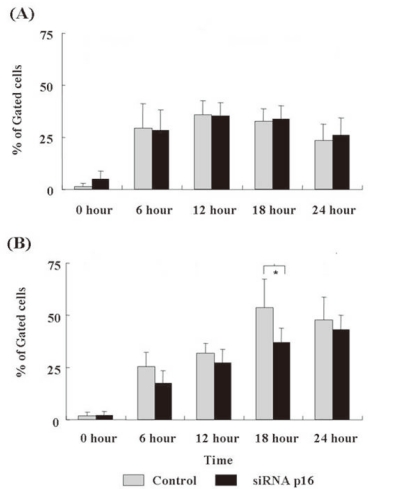
3. Apoptotic changes after DX treatment
We assessed the time-dependent apoptotic changes after DX treatment using Annexin V/PI staining of cells by flow cytometry. After the DX treatment, both p16+ and p16- NC-37 cells showed a marked initial increase in Annexin V-stained cells (the early apoptotic cells) at 6 h, followed by a decrease at 24 h (both P<0.05). However, there was no statistical difference between the groups.
The late apoptotic cells (double-positive cells) in p16- NC-37 cells increased through 6~18 h and reached a maximum at 18 h, and >50% of the cells were apoptotic. In contrast, the late apoptotic cells increased in a time-dependent manner over 24 h in p16- NC-37 cells. Overall, p16+ NC-37 cells was more susceptible to DX-induced late apoptosis than p16- NC-37 cells, and the result at 18 h was significant (P<0.05, Fig. 3).
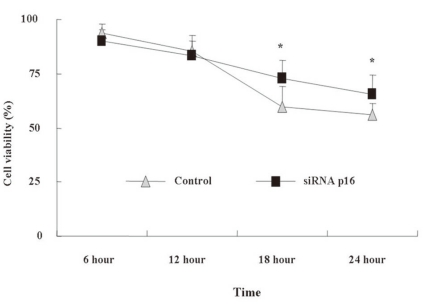
4. Time-dependent cell viability after DX treatment
We assessed time-dependent cell viability using the AB assay. AB is a redox indicator that produces a colorimetric change in the fluorescent signal in response to metabolic activity. Within 12 h, the cell viability between p16+ and p16- NC-37 cells was not significantly different. At 18 and 24 h, the cell viability was reduced compared to the value at 12 h in both groups (P<0.05), and the degree of the decrease was more severe for p16+ NC-37 (P<0.05, Fig. 4).
Discussion
Clinically, GC resistance indicates a poor prognosis in ALL treatment1-3). However, the molecular mechanisms of the anti-leukemic effects and GC resistance are not clear3-5). It has been hypothesized that GC induces apoptosis by affecting the Bcl-2 gene directly or inducing the release of apoptogenic factors. In addition, GC-induced cell cycle arrest in leukemic cells may be independent of apoptosis induction and associated with increased expression of CDK inhibitors such as p273-5). Factors affecting GC sensitivity include the availability of the hormone, tissue-specific factors, the intracellular metabolism of the hormones, and GR responses7). There is increasing evidence that the level of GR expression auto-induced by GC is associated with GR molecular functions, including steroid responsiveness and resistance5, 14, 15).
The inactivation of p16 has been confirmed to varying degrees in T-cell leukemias and other hematologic malignancies16-23), and INK4A gene knockout mice develop spontaneous lymphomas at high frequency24). These findings suggest that p16 is involved in the negative regulation of tumor proliferation and progression in the lymphoid lineage. Recently, p16 was also studied as a prognostic factor for the diagnosis of hematologic malignancies3, 13). It was reported that the inactivation of p16 might be associated with the presence of minimal residual disease at the induction of chemotherapy, and loss of p16 function might also be a marker of chemoresistance in non-high-risk B-cell ALL3). However, other studies of several hematologic malignancies do not confirm these findings11). Ausserlechner et al reported that p16 sensitizes lymphoblastic leukemia cells to apoptosis by GC14). Using p16 gene transfection to GC-sensitive T lymphoma cell lines, they observed that after GC treatment the p16+ cells showed increased GR expression and sensitivity to GC, whereas the p16- cells did not. They postulated that because leukemic cells may have a program for escaping apoptosis by suppressing apoptotic pathways, p16 inactivation might be required for lymphoid malignancies to escape from as yet unrecognized tumor surveillance, including GC-induced apoptosis14). However, the mechanisms of p16 inactivation contributing to GR regulation and subsequent apoptosis remain unknown.
Few studies have examined the relationship between GC responsiveness and p16. In this study, we evaluated the time course of GC-induced GR expression changes and the effect of p16 on GC-induced apoptosis using p16 siRNA transfection of a B-cell lymphoblast cell line, NC-37 cells. We found a pattern of intracytoplasmic GR expression after DX treatment within 24 h. That is, the initial GR levels peaked at 18 h, followed by a sudden decrease at 24 h in p16+ and p16- NC-37 cells; the former tended to show higher expression rates (Fig. 2). The repression of GR expression 24 h after GC treatment has also been observed in other studies25-27). Along with GR expression, similar patterns of early apoptotic cells induced by DX were observed in both groups (Fig. 3A). However, late apoptotic cells increased in a time-dependent manner, and this was more marked at 18 h for p16+ NC-37 cells. This suggests that p16 influences DX-induced late apoptosis more than early apoptosis (Fig. 3B). The viable cell assay showed similar results for both groups (Fig. 4).
Combined, our results suggest that p16 is positively correlated with GR expression and GC-induced late apoptosis. Further effort is necessary to identify additional genetic changes in cell cycle regulators to complement these findings. Studies of p16 and GC responsiveness may lead to new treatment modalities, such as a combination of GC with substances mimicking p16 function, including CDK4- or CDK6-inhibiting peptides, for hematologic malignancies that do not express p1614). In addition, because GR expression is not the sole factor determining GC sensitivity4-6), further studies must examine the relationship between GC-induced apoptosis and the interaction of other signaling events associated with p16 or other proteins.
In conclusion, although p16 has a role in GR expression and apoptosis induced by GC, it remains to be seen whether the GC-induced apoptosis mechanism can interact with the other signaling events associated with the p16 gene operating during such malignant changes. This observation might have important implications for cancer therapy.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


