

Introduction
Ornithine transcarbamylase (OTC) deficiency is the most common inborn error of the urea cycle with resulting hyperammonemia, which is a medical emergency in newborns1). Catabolism of amino acids results in the production of free ammonia which is highly toxic to the central nervous system. Ammonia is detoxified to urea through pathways known as the urea cycle. The related enzymes are carbamyl phosphate synthetase, OTC, argininosuccinic acid synthetase, argininosucci nic acid lyase and arginase2).
OTC is functioning in process ornithine and carbamyl phosphate to the citrulline synthesis. OTC defects cause accumulation of the precursors to ammonia, glutamate, glutamine, and alanine and decrease the serum arginine and citrulline. We can also detect an increase in urinary orotic acid.
OTC deficiency is known to have X-linked inheritance. OTC gene is located at Xp 21.1 and cDNA was cloned in 19843). In 1988 structure of the OTC gene was found4). Clinical features are vomiting, hypotonia, lethargy, seizures and coma5). Such neurologic symptoms are mainly due to the accumulation of glutamate in the central nervous system as a result of interruption of the normal ammonia breakdown process via the urea cycle. In classic cases, male newborn patients present with severe hyperammonemia in the first few days of life5).
We encountered a male newborn patient who had acute severe hyperammonemia in his third day of life. He was diagnosed with OTC deficiency through serum amino acid and urine organic acid analysis and OTC deficiency gene mutation. This is one of the few reported cases in which ammonia was effectively removed by continuous veno-venous hemodiafiltration in a newborn.
Case report
A male patient was born weighing 3,010 gm by normal spontaneous vaginal delivery at 40 weeks and 5 days of gestational age. Apgar score was 7 and 9 at 1 and 5 minutes, respectively. The parents were not in a consanguous marriage and had no medical problems except Bechet's disease in the mother. The patient was in good condition in the first 2 days of life with tolerable oral feeding.
In his 60th hour of life, the baby was admitted into neonatal intensive care unit (NICU) due to lethargy with moaning sound, heavy sweating, hypothermia and poor feeding. On the admission day, the infant had a seizure for 2 minutes with lip smacking and apnea. The patient was intubated and put on a ventilator. He was started on an anticonvulsant (Phenobarbital). The patient's serum ammonia level was above 1,700 µg/dL.
Other laboratory findings upon admission were as follows: Leukocytes 21,890/µL, hemoglobin 7.8×106/µL, hematocrit 76%, platelet 279,000/µL, prothrombin time 83% (international normalized ratio, 1.13), activated partial thromboplastin time 16.6 seconds, C-reactive protein 4.17 mg/dL, serum glucose 130 mg/dL, total bilirubin 8.3 mg/dL, direct bilirubin 1.0 mg/dL, aspartate aminotransferase 55 IU/L, alanine aminotransferase 25 IU/L, blood urea nitrogen 6.7 mg/dL, creatinine 1.14 mg/dL and lactate dehydrogenase 549 IU/L. The patient's serum electrolytes were: sodium 153 mmolL/L, potassium 6.0 mmol/L and chloride 116 mmol/L. The arterial blood gas analysis showed pH 7.2, pCO2 58 mmHg, pO2 90 mmHg, bicarbonate 21.9 mmol/L, O2 saturation 95%, ionized calcium 2.82 mg/dL (4.5 to 5.2), ionized magnesium 0.88 mg/dL (1.09 to 1.46) and lactate 5.6 mmol/L (0.5 to 1.6).
Continuous veno-venous hemodiafiltration was immediately initiated to eliminate excess ammonia.
In plasma amino acid analysis, several amino acids were found at slightly to moderately high levels: methionine 94.1 nmol/mL, tyrosine 255.2 nmol/mL, lysine 613.7 nmol/mL, proline 581.6 nmol/mL, alanine 1,099.8 nmol/mL and glutamine 1,450.3 nmol/mL. However, the citrulline peak was not detected. In plasma organic acid analysis, lactate was 3,934 µmoL/L, while pyruvate and fumarate were 365 µmoL/L and 6.12 µmoL/L, respectively. Urine organic acid analysis showed that orotic acid increased markedly.
Neurosonography revealed diffuse heterogenous increased echogenicity in the white matter of the brain parenchyma, and the abdominal sonography showed no abnormalities. The electroencephalogram suggested moderate to severe encephalopathy.
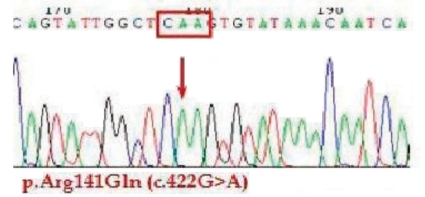
A genetic study confirmed the diagnosis of OTC deficiency. A single nucleotide change from G to A was discovered (c.422G>A) and this change resulted in missesnse mutation, aminoacid change from arginine to glutamine at codon 141 (p.Arg 141 Gln) (Fig. 1).
We suspected the urea cycle defect, and all dietary and intravenous nitrogen intakes were discontinued. The patient started continuous renal replacement therapy (CRRT) with the mode of continuous veno-venous hemodiafiltration in the 3rd day and continued for 7 days. The serum ammonia level decreased rapidly (Fig. 2).
CRRT was performed with the Prisma and M10 filter kit (Gambro, Meyzieu, France). Hemosol B0 (Gambro Lundia AB, Lund, Sweden) containing Na+ 140 mmol/L, Cl-109.5 mmol/L, Ca2+ 1.75 mmol/L, Mg2+ 0.5 mmol/L, lactate 3 mmol/L, HCO3- 32 mmol/L) was used as the dialysate and the replacement solution.
The CRRT was primed with normal saline solution (1 L) mixed with heparin (5,000 IU). Vascular access was obtained through the right internal jugular vein and a broviac catheter (6.5 Fr.) was inserted. To maintain the activated clotting time between 170 and 210 seconds, heparin (0 to 20 IU/kg/hr) was administered according to the patient's coagulation status. The ultrafiltration rate was modulated by fluid intake and diuresis. The blood flow rate ranged between 20 and 25 mL/min. The flow rate of the replacement solution and the dialysate was 150 mL/hr.
The serum ammonia level decreased rapidly to 422 µg/dL after 24 hours of dialysis, to 222 µg/dL after 57 hours of dialysis, and finally to 200 ug/dL (Fig. 2). The patient maintained stable vital sign without hypotension and electrolyte imbalance during CRRT was processing. There was no catheter related complication, also.
CRRT was discontinued, and extubation was done on the 5th day of dialysis. However the patient was kept on CRRT for three more days due to a subsequent re-elevation of the ammonia level to 364 µg/dL. After discontinuing CRRT, the patient started a protein-free diet, and oral buphenyl (50 mg) and oral sodium benzoate were supplied. Serum ammonia was not elevated for about twenty days after stopping CRRT.
A lactulose enema was done every day. Protein was reintroduced into the patient's diet one week after stopping the dialysis. We gradually increased protein portions in his diet while he was in NICU for close monitoring and conservative care. The patient had no additional seizures and was stable in vital signs. However, his general condition got worse in about 20 days after stopping CRRT. The patient was malnutrition state and showed skin lesion resembling acrodermatitis enteropathica. He showed liver function deterioration with hypoalbuminemia and serum ammonia level began to rise slightly. Furthermore he suffered candida infection in urinary tract and could not overcome the septic condition. Finally the patient expired because of fungal infection and septic shock.
Discussion
We have presented one of the most severe manifestations of OTC deficiency. Abrupt mental deterioration occured in the few days of life of a healthy male newborn patient, beginning with oral feeding1). Mild forms are commonly seen in heterozygous females and in some affected males, usually after infancy6). We suspected an inherited metabolic disorder, especially a defect of the urea cycle because of the typical clinical course and extremely high ammonia level. It took several days to diagnose OTC deficiency because of the time taken to analyze plasma amino acids and urine organic acids. However, if a urea cycle defect is suspected, before the final diagnosis is confirmed, emergency treatment of acute hyperammonemia should be started. In this case, mutation of OTC gene was also confirmed. This mutation (p.Arg 141 Gln) is known as most common point mutation in OTC deficiency occuring at CpG hot spot and it shows no residual enzyme activity7,8).
The principles of treatment include the following9). 1) elimination of protein intake and minimization of protein catabolism, 2) administration of substrates of the urea cycle that are lacking as a consequence of enzyme defect 3) removal of ammonia and 4) treatment of other conditions such as shock, sepsis and seizures. Stopping protein intake and providing sufficient calories in the form of glucose and lipids is important. Protein intake should be restricted to 0.5 to 2.0 g/kg per day. Because urea cycle defects lead to citrulline and arginine deficiency, supplementing these can replenish urea cycle substrates10,11). This permits the urea cycle to function and enables it to remove additional nitrogen waste, while supplements of essential amino acids reverse proteolysis. Ammonia removal is made possible by the administration of sodium phenylacetate and sodium benzoate that use alternative pathways to excrete ammonia10). Phenylacetate conjugates with glutamine and synthesizes phenylacetyl glutamine. Benzoate conjugates with glycine and synthesizes hippurate. Phenylacetyl glutamine and hippurate can be eliminated in urine. In mildly affected patients, restriction of nitrogen and activation of alternative pathways may be sufficient. In severe cases, however, dialysis is needed to rapidly remove serum ammonia.
Various dialysis modalities have been used to remove ammonia. They include peritoneal dialysis (PD), intermittent hemodialysis (HD), continuous veno-venous hemodialysis, and continuous arteriovenous hemodialysis. Previous hyperammonemic patients have been treated with peritoneal dialysis or exchange transfusion but peritoneal dialysis has shown slow removal rates. HD is about 10 times more effective than PD in the clearance of toxins with low molecular weight such as ammonia12,13).
In a recent trial, continuous veno-venous hemodialysis showed faster reduction of ammonia and better long term prognosis than continuous peritoneal dialysis14). Comparing HD and CRRT, HD is 2 to 3 times more effective in the clearance of ammonia13,15), but many recommend CRRT over HD in treatment of neonatal hyperammonemia, because HD can cause hemodynamic instability and hypotension in small infants14,16-18).
Further controlled trials are required to compare HD and CRRT in efficacy, occurrence of complications and prognosis.
In our case, continuous veno-venous hemodiafiltration was successful in decreasing serum ammonia level and maintaining stable vital signs without any electrolyte imbalance or other problems, and this case is the first successful treatment of hyperammonemia by CRRT in our hospital. Because the application of CRRT to a newborn is not technically available in all tertiary care centers in Korea, rapid transportation to a center where this is available would be essential and may reduce hyperammonemia-induced brain injury.
However, he later expired because of sepsis. For prolonged survival, providing sufficient nutrition and calories, including some degree of amino acids, is important. This is because protein and energy are necessary for growth, life support and preventing metabolic disturbance. This patient could not maintain a healthy nutritional state and suffered from deterioration of hepatic function as the protein increased in his diet, and had difficulties in overcoming his septic condition.
For definite treatment, liver transplantation is necessary in patients who have been well controlled in hyperammonemia. We need more advanced therapeutic strategies such as orthotopic liver transplantation, liver cell transplantation or gene therapy.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


