


Introduction
In parallel with nephrogenesis, renal vascularization takes place in a tightly regulated pattern1). Precise and timely assembly of the kidney vessels with their respective nephrons is an essential morphogenetic event that establishes a functioning kidney required for independent extrauterine life2). The renin-angiotensin system (RAS) plays a vital role during kidney growth and development. Numerous studies by using gene targeting or pharmacologic interruption of the RAS have shown its importance in nephrovascular development3,4,5). Pharmacological inhibition of angiotensin II actions in immature kidneys causes significant alterations in renal architecture, such as immature glomeruli and papillae, dilated tubuli, and arrested vascular development6,7,8).
Microvascular rarefaction after kidney injury is implicated in hypoxia, chronic inflammation, and progressive loss of renal function9,10). Experimental data demonstrate that the degree of glomerular and peritubular capillary loss is related with the severity of glomerulosclerosis and interstitial fibrosis11). Reciprocal signaling between capillary endothelial cells and pericytes supports capillary integrity12), and the vital cross talk between the 2 cell types is central to injury responses in the kidney13). During arteriolar development, pericytes trigger endothelial cell proliferation and migration. Endothelial cells, in turn, stimulate the pericyte precursor cell population14). Various signaling pathways, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) signaling system, play an important role for these paracrine interactions2). VEGF-A signaling from pericytes to endothelial cells is involved in developmental and pathological angiogenesis15,16,17,18). The VEGF receptor 1 (VEGFR1) and VEGF receptor 2 (VEGFR2), organize these processes of angiogenesis19). PDGF-B is also implicated in vessel formation and glomerulogenesis during fetal life, as revealed by microaneurysm development in capillary beds and the loss of mesangial cells in PDGF-B null mice6). PDGFR-β signaling is crucial to the recruitment of pericytes or perivascular progenitor cells20).
Several lines of evidence indicate that angiotensin II is a regulator of angiogenesis in kidney development, as well as various disease settings6,21,22). A previous study demonstrated that short-term exposure to pressor doses of angiotensin II caused microvascular injury with peritubular capillary loss23). Despite the well-known renal abnormalities after neonatal RAS blocking, mechanisms that govern the development of the renal vasculature remain unclear. Therefore, we investigated whether neonatal angiotensin converting enzyme (ACE) inhibition causes inappropriate regulation of angiogenesis with capillary endothelial injury. The molecular events that maintain the renal vasculature were also examined through the signaling pathways of the VEGF/VEGFR and PDGF-B/PDGFR-β system after RAS blockade in developing rat kidney.
Materials and methods
1. Animal preparation
Twenty-four neonatal rat pups from two pregnant Sprague-Dawley rats were breastfed by their own mother throughout the study. From birth, body weights were measured daily, and 30 mg/kg of enalapril (Sigma Chemical Co., St. Louis, MO, USA) (n=14) or vehicle (control group, n=10) were administered daily by via an orogastric tube. The rats were sacrificed at 8 days of age. Their kidneys were harvested and processed for the study. One whole kidney (right kidney) from each rat was used for light microscopy and immunohistochemistry, and the other whole kidney (left kidney) for Western blot. The experimental protocol was approved by the Animal Care Committee of Korea University Guro Hospital.
2. Histologic examinations
Harvested kidneys were treated in 10% formalin solution (Sigma Chemical Co.) and embedded in paraffin. The samples were then cut into 4-µm sections and dried on siliconized slides (Muto-Glass, Tokyo, Japan). The slides were deparaffinized with xylene solution, followed by staining with hematoxylin (Gill No. 2, Sigma Chemical Co.) for nucleus and eosin (Sigma Chemical Co.) for cytoplasm.
3. Western blotting
The extracted proteins were solubilized in 5× sodium dodecyl sulfate (SDS) loading buffer for 5 minutes at 95℃ and separated by electrophoresis on 10% SDS-polyacrylamide gels under reducing conditions. Equal amounts of 5–15 µg of proteins were loaded per lane. Then, the proteins were transferred to nitrocellulose membranes (KPL, Gaithersburg, MD, USA). The membranes were blocked in 5% skim milk with Tris-buffered saline and tween 20 (TBS-T) (0.05% Tween 20 in 50mM of Tris, 150mM of NaCl, and 0.05% NaN3 [pH 7.4]) at room temperature for 1 hour. The membranes were washed 2 times in TBS-T and incubated for 18 hours at 4℃ with rabbit polyclonal antiserum VEGF-A (dilution 1:150; Santa Cruz Biotechnology, Santa Cruz, CA, USA), VEGFR1 (dilution 1:200; Santa Cruz Biotechnology), VEGFR2 (dilution 1:200; Cell Signaling Technology, Danvers, MA, USA), PDGF-B (dilution 1:100; Abcam, Cambridge, UK), PDGFR-β (dilution 1:500; Cell Signaling Technology), and CD31 (dilution 1:200; Santa Cruz Biotechnology). Thereafter, the membranes were washed 2 times with TBS-T and incubated for 40 minutes with an antirabbit IgG (GE Healthcare, Buckinghamshire, UK) and an antimouse IgG (GE Healthcare, Buckinghamshire, UK) at room temperature. To control for equal loading, β-actin (dilution 1:1,000; Cell signaling technology) and antimouse IgG conjugated horseradish peroxidase (dilution 1:1,000; Cell signaling technology) were used as primary and secondary antibodies with the same method as described above. The developed x-rays were scanned using the Epson GT-9500 (Seiko Corp., Nagano, Japan) and the results were quantified by densitometry (Image PC alpha 9, National Institutes of Health, Bethesda, MD, USA).
4. Immunohistochemistry
For assessing expression, five kidneys in each group were selected for representative immunohistochemical staining, using an avidin-biotin immunoperoxidase method (Vectastain ABC kit, Burlingame, CA, USA). Immunohistochemistry was performed on paraffin sections as described previously3). Primary antibodies against VEGF-A (dilution 1:100; Santa Cruz Biotechnology), VEGFR1 (dilution 1:20; Santa Cruz Biotechnology), VEGFR2 (dilution 1:150; Cell Signaling Technology), and CD-31 (dilution 1:20; Santa Cruz Biotechnology) were used.
5. Quantitative analysis of capillary density
The loss of glomerular and peritubular capillaries was assessed after immunostaining for CD31 using a point detection method. Positively stained CD31 areas in the renal cortex were identified by point detection, and the average CD 31 staining in at least 20 fields of cortex per kidney section from each of 5 rats was determined at a magnification of ×400. Histological quantification was analyzed in a blinded fashion using a 400-fold magnification.
Results
1. Renal histological alterations
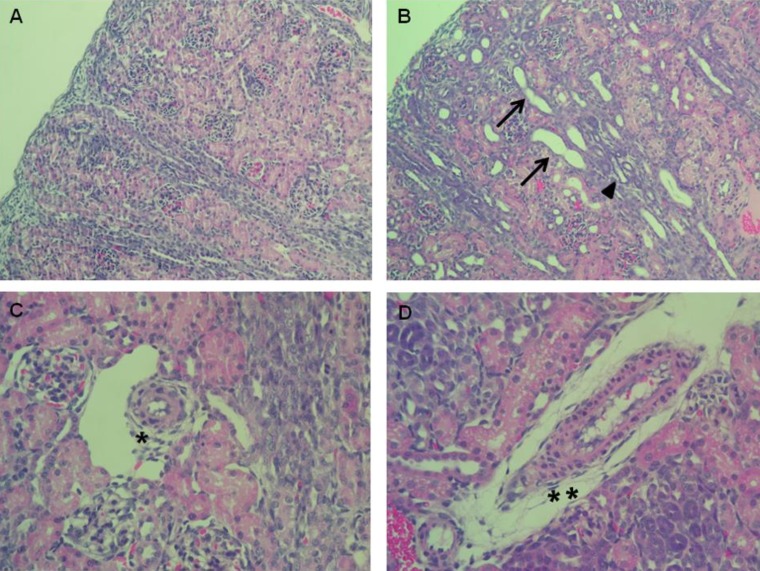
Enalapril-treated rat kidneys showed extensive tubular and vascular abnormalities, as compared to the control kidneys (Fig. 1A–D). Hematoxylin and eosin staining of the kidney indicated that enalapril-treated rats exhibited dilated and atrophic tubules, as compared with the control group (Fig. 1A, B). Disrupted intrarenal arterioles were found in enalapril-treated but not control kidneys (Fig. 1C, D).
2. Changes in capillary density
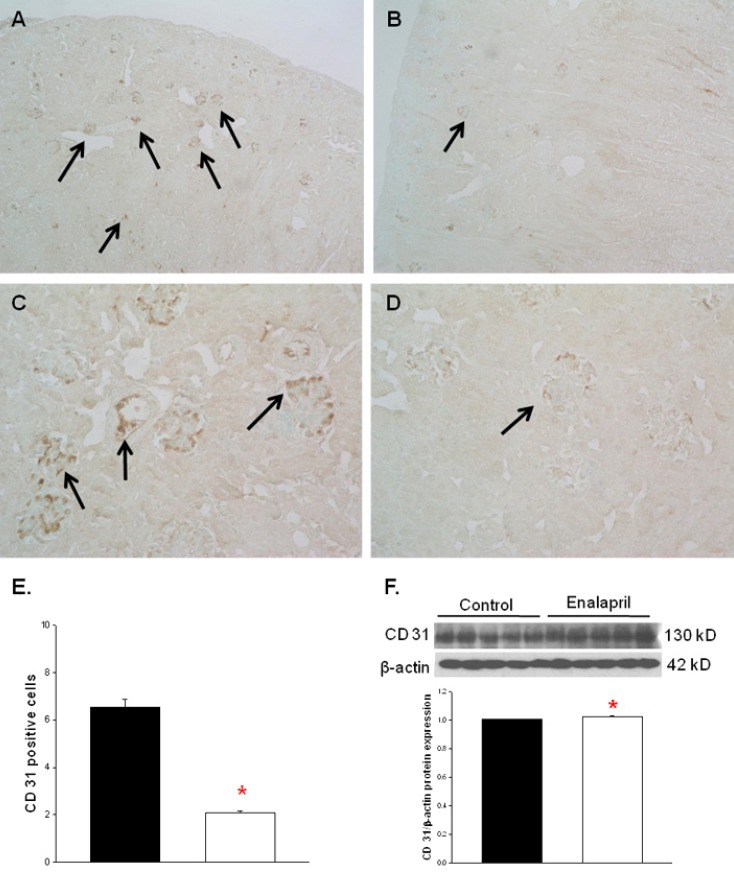
The capillary endothelial marker CD31 was easily and accurately identified in normal kidney tissues (Fig. 2A, C). However, there was a visible decrease in both glomerular and tubulointerstitial CD31 staining in enalapril-treated kidneys (Fig. 2B, D). The cortical capillary densities, detected with CD31 immunohistochemical staining, were significantly decreased in the enalapril-treated group, as compared with the control group (P<0.05) (Fig. 2E). In contrast, the immunoblots of total kidney showed that the CD31/β-actin protein expression was enhanced in enalapril-treated groups, as compared to the control group (P<0.05) (Fig. 2F).
3. VEGF, VEGFR1, and VEGFR2 expression
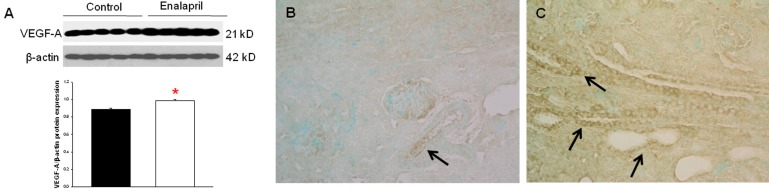
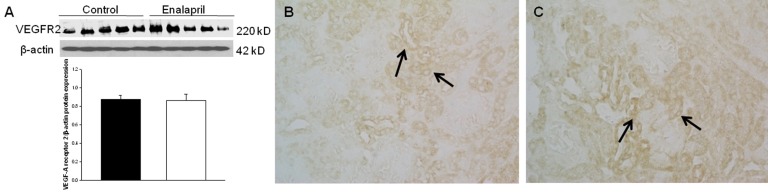
In the enalapril-treated group, intrarenal VEGF-A expression was increased, as compared to the control group (Fig. 3A–C). The immunoblots showed that the VEGF-A/β-actin protein expression was significantly increased in the enalapril-treated group (P<0.05) (Fig. 3A). Immunohistochemically, VEGF-A expression was detected within some glomeruli and tubular epithelial cells in the control group (Fig. 3B). It was more strongly detected in several dilated tubules and glomeruli in the enalapril-treated group (Fig. 3C). In comparison, intrarenal VEGFR1/β-actin protein expression was reduced in the enalapril-treated groups, as compared to the control group (P<0.05) (Fig. 4A). Immunohistochemically, VEGFR1 expression was detectable within some of the tubular epithelial cells and glomeruli in the cortex of the control kidneys (Fig. 4B); however, it was rarely detected in tubules and glomeruli in the enalapril-treated group (Fig. 4C). There was no difference in VEGFR2/β-actin protein expression between the enalapril-treated group and the controls (Fig. 5A). The immunohistochemical staining showed that VEGFR2 expression was similarly detectable in cortical tubular segments of both the control and enalapril-treated groups (Fig. 5B, C).
4. PDGF-B and PDGFR-β expression
The expression of PDGF-B and PDGFR-β was not different between the two groups. In the enalapril-treated group, PDGF-B/β-actin protein expression was decreased and PDGFR-β/β-actin protein expression was mildly increased; however, there were no statistical significances (Fig. 6A, B).
Discussion
In the present study, enalapril treatment caused tubular dilatation and atrophy and vascular abnormalities in the neonatal rat kidney. In the enalapril-treated kidneys, the cortical capillary density decreased as detected with CD31 immunohistochemical staining, while the capillary endothelial cell protein level of total kidney, measured by CD 31 Western blots, increased. Intrarenal VEGF-A protein expression was significantly higher, whereas VEGFR1 protein expression was lower in the enalapril-treated group, as compared to the control group. Enalapril treatment had no effects on the expression of VEGFR2, PDGF-B, and PDGFR-β. These findings indicated that the alteration in the local expression of both angiogenic and antiangiogenic factors in the kidney may mediate the impaired capillary repair, contributing to renal injury after the blockade of the RAS in the developing rat kidney.
Inhibition of the RAS in animals with fetal and neonatal kidneys results in striking renal histological alterations, suggesting a vital role for this system in renal development1,3,4). Renal histological abnormalities are typically characterized by papillary atrophy, interstitial inflammation and fibrosis, tubular dilatation and atrophy, and renal vascular changes1,7). Blockade of angiotensin II action in rats during the first 12 days after birth, when arteriolar branching is at its peak, produces remarkable deterioration of renal vascular development identified by fewer, shorter, and thicker radial and afferent arterioles. This occurs with decreased glomerular size and tubular atrophy with attendant reduction in entire kidney growth5). Consistent with these findings, the present study confirmed that treating rats with enalapril for the first 7 days of postnatal life exhibited severe renal structural impairment, including tubular dilatation and atrophy and abnormal increase in the size of the vascular wall. Capillary endothelial repair, detected with immunohistochemical staining and immunoblotting, appeared impaired. Although the immunoblots of total kidney showed that the CD31 protein expression was enhanced in the enalapril-treated groups, the cortical capillary densities, measured by CD31 immunohistochemical staining, were significantly reduced in enalapril-treated rats.
Preservation of the microvasculature is important for the delay of progression of kidney disease11). In the healthy kidney, the close reciprocal signaling between pericytes and capillary endothelial cells supports capillary integrity12). Capillary loss, distinguished by the destruction of the microvasculature, is a characteristic of progressive kidney disease and corresponds closely with the degree of renal function decline11,24). Numerous studies found that, although there is an initial proliferative response of the capillary endothelium in response to renal injury, proliferation is not maintained, and a progressive capillary loss develops in various models of renal disease25,26). Morphometric quantification of the renal microvasculature from day 2 after unilateral ureteral obstruction (UUO) injury, demonstrated increased capillary density subsequent to an initial insult, revealing the early angiogenic response to injury; however, in response to continuous insults, there was marked loss of the microvasculature by day 10 after UUO injury, implying that endothelial cell proliferation may be initially effective but later impaired13). In the present study, we observed that enalapril treatment after birth stimulated the capillary endothelial cell protein level of whole kidney by postnatal day 8. In contrast, CD31 positive-endothelial cell immunolocalization in the renal cortex was suppressed. It is likely that the endothelial proliferative response may initially occur in the entire kidney, but, at the same time, the cortical capillary loss can be ongoing in this experimental model. The loss of microvasculature can be mediated in part by a decline in early endothelial cell proliferation, and the impaired capillary repair is modulated by changes in the expression of both angiogenic and antiangiogenic factors11).
VEGF-A, a major angiogenic factor, is essential for supporting peritubular and glomerular capillary integrity in the healthy kidney. In renal disease, however, VEGF-A can exert both beneficial and detrimental roles27). VEGF-A binds its 2 receptors, VEGFR1 and VEGFR2, that mediate physiological and pathological angiogenesis19). VEGFR1 has a dual role that involves a negative regulatory effect on angiogenesis by trapping VEGF-A in the embryo, and a positive angiogenesis regulation in adulthood in a tyrosine kinase-dependent manner19,28). It is also involved in the inflammatory response by provoking macrophage chemotaxis, vascular permeability, and vessel stabilization29,30). VEGFR1 acts mainly as a coreceptor, having a high affinity for VEGF but a lesser signaling properties11). In contrast, VEGFR2 has a lesser affinity for VEGF but displays much more intracellular kinase activity upon binding. It functions as a direct signal transducer in the process of pathological angiogenesis19,31). Notably, the renal disease found in mice with podocytes lacking VEGF is similar to the renal disease detected in adult mice lacking VEGFR2, with obvious glomerular endothelial cell defects, reflecting that VEGFR2 plays a key role in capillary endothelial cell preservation. Conversely, postnatal deletion of VEGFR1 does not cause a phenotype similar to the loss of VEGF from podocytes, supporting that VEGF signaling through VEGFR2 is critical in the glomerular microvasculature32). In the present study, we showed that a key angiogenic factor VEGF-A expression was enhanced, and its major angiogenic receptor VEGFR2 was unchanged after enalapril treatment by postnatal day 8. VEGFR1, which plays a negative role in angiogenesis in the early development19), was reduced in enalapril-treated rat kidneys. These findings indicated that ACE inhibition in neonatal rats can induce early angiogenic response, along with increased expression of VEGF-A and decreased expression of VEGFR1, within the tubular epithelium and glomerular microvasculature. However, the lack of signaling through VEGFR2 may impair the angiogenic environment and eventually induce capillary regression.
Parallel changes were also found in the PDGF signaling of the present study. We confirmed that enalapril treatment for the first 7 days after birth had no effects on the expression of pro-angiogenic factor PDGF-B and its receptor PDGFR-β in developing rat kidneys. PDGF-B is released from endothelial cells, which promotes pericyte proliferation by binding to PDGFR-β2). Regulated PDGF-B/PDGFR-β signaling is crucial in vascular stabilization and in sprouting angiogenesis20,33,34). Kidneys deficient in PDGF-B or PDGFR-β do not develop mesangial cells (a subtype of pericytes) and have anomalous glomerular capillaries20). Interestingly, in the UUO model of renal injury, VEGFR2 blockade prevented signaling in pericyte PDGFR-β as well as at the endothelial cell VEGFR2. Both VEGFR2 blockade and PDGFR-β blockade markedly attenuated fibrosis and capillary rarefaction during progressive kidney injury. The vital cross talk between endothelial cells and pericytes is central to the injury responses in the kidney, and both the VEGF-A/VEGFR2 and PDGF-B/PDGFR-β signaling play key roles in this endothelium-pericyte crosstalk13). Considering these findings, we speculate that injured capillary endothelium in our experimental model becomes susceptible to VEGF-A/VEGFR1 signaling, which triggers an angiogenic response initially. However, angiogenic signaling through VEGFR2 might be ineffective and, in parallel, PDGF-B/PDGFRβ signaling can be unresponsive to renal injury by day 8 after RAS blockade. For angiogenesis to function suitably, a cooperated interplay of several key players is required35). The results of our study suggested that renal injury after RAS blockade in immature kidney is partially due to an impaired angiogenic response, which may be further aggravated by the additional induction of an antiangiogenic environment27).
Our study had some limitation. Deterioration of the kidney vasculature was not quantified, except for capillary endothelium measures using CD31. Further quantitative data, such as length, volume, and/or surface area of kidney vasculature in both the cortex and the medulla would strengthen the results of our data. Additional analysis of antiangiogenic molecules may further improve our findings. We should also consider that ACE inhibition exerts beneficial or undesirable effects on capillary repair, dependent on time and predominant pathology.
Taken together, our findings showed that the RAS is important for the preservation and/or development of renal vessels and tubules. Enalapril treatment for 7 days after birth in neonatal rats induced the deterioration of kidney microvasculature with improper VEGF-A/VEGFR signaling. The capillary endothelial repair seemed impaired. Because kidney disease is accompanied by the degeneration of renal vasculature and/or the reappearance of embryonic pathways, identifying the fundamental mechanisms for the formation and preservation of the renal microvasculature can be a strategy to protect renal structure and function2). Further studies will be required to define the mechanisms that yield functional significance to the abnormal vascular growth after RAS blockade in developing kidney.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


