


Introduction
Pulmonary alveolar proteinosis (PAP) is a rare type of diffuse lung disease characterized by intra-alveolar accumulation of pulmonary surfactant. Histopathologic examination shows that, in patients with this condition, the distal air spaces are filled with a granular, eosinophilic material that stains positively with periodic acid–Schiff reagent and is diastase resistant. This material contains large amounts of surfactant proteins and lipids. It is suspected that the primary mechanism of these accumulation is impaired catabolism by alveolar macrophages1).
The natural history of primary PAP is highly variable, making prognostic and therapeutic decisions difficult. Currently, whole lung lavage (WLL) is the treatment of choice for PAP. Although WLL can lead to prolonged remission of PAP in adults, some children may demonstrate relapse2,3). We here present a pediatric case of PAP diagnosed by lung biopsy that was incompletely treated by WLL. This is the first childhood case of PAP relapse despite WLL in Korea.
Case report
An 11-year-old boy was admitted to the hospital because of dyspnea on exertion. He first presented with dry cough, 6 months prior to the current admission. He developed symptoms of dyspnea on exertion and had trouble in climbing stairs. He presented with nausea and intermittent vomiting, which gradually developed and progressed.
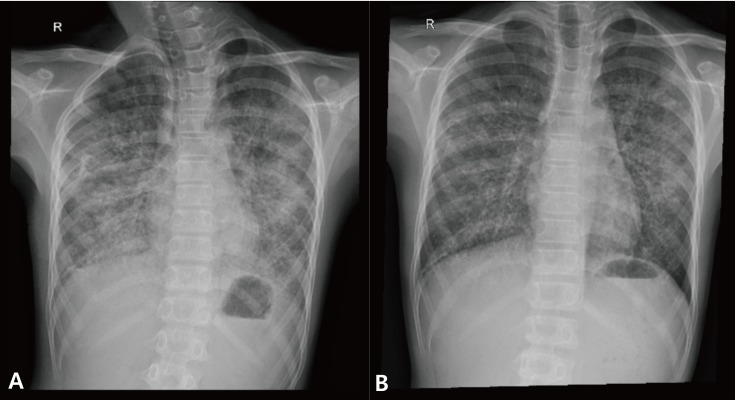
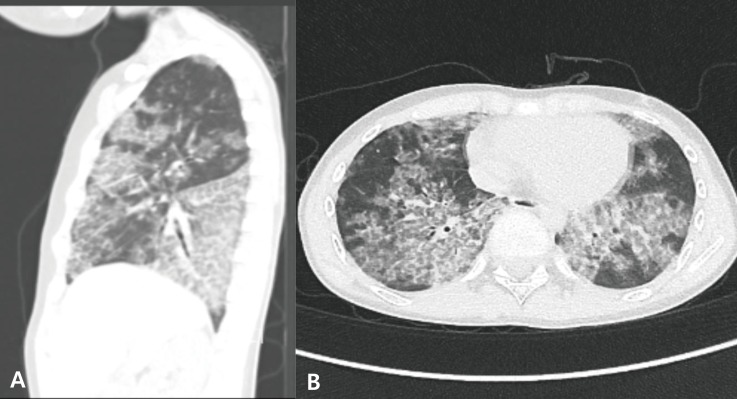
He first visited our institute to rule out gastroesophageal reflux. During the physical examination, the chest wall expanded symmetrically without retraction and breathing sounds were clear without crackle or wheezing. Heartbeats were regular without audible murmur. The abdomen was soft and flat with normoactive bowel sounds. There was neither tenderness nor rebound tenderness on the abdomen. The liver and spleen were not palpable. Since he also complained of intermittent dry cough and dyspnea on exertion, he underwent chest radiograph, which revealed bilateral symmetric ground-glass opacities with relative sparing of the apices (Fig. 1). The upper gastrointestinal series showed diminished bowel motility without evidence of gastroesophageal reflux, which implied the unlikelihood of recurrent aspiration. Chest computed tomography (CT) scan demonstrated geographic areas of ground-glass opacity and septal thickening with a “crazy-paving” appearance (Fig. 2).
To make a definitive diagnose, he underwent video-assisted thoracoscopic lung biopsy; the histopathological examination revealed diffuse eosinophilic material in alveolar spaces, which was consistent with PAP (Fig. 3). Since his dyspnea progressed to the extent that he had difficulties in his daily activities, he was finally admitted to our institute to undergo bilateral WLL.
He had no family history related to pulmonary or autoimmune problems. His heart rate was 129 beats/min and respiratory rate was 26 breaths/min and His transcutaneous oxygen saturation while breathing room air was 92%. Venous blood gas analysis on room air revealed a pH of 7.42, PCO2 39 mmHg, PO2 42 mmHg and HCO3
- 25.3 mEq/L. His total leukocyte count was 6,440/mm3 and his hemoglobin level was 16.1 g/dL. The C-reactive protein level was 0.16 mg/dL. The results of renal function and liver function tests were normal. Investigations for his immunologic work-up showed IgG, IgA, and IgM levels to be within the normal range. His pulmonary function tests revealed a restrictive pattern, with a forced vital capacity (FVC) of 0.69 L (26% of predicted value), a forced expiratory volume in 1 second (FEV1) of 0.59 L (24% of predicted value), and a ratio of FEV1/FVC 86%.
He was subjected to therapeutic WLL of alternate lungs, one each on 2 separate days. Before the procedure, adequate isolation of the right and left lungs was ensured. The procedure was first performed in the right lung while ventilating the left lung using a double lumen endotracheal tube. He was positioned to the left lateral decubitus. Aliquots of 500–600 mL, roughly equivalent to tidal volume, of buffered normal saline, warmed to body temperature, were instilled into the lung. The fluid was allowed to flow into the lungs by gravitational force until the flow of solution ceased. After each instillation, the fluid was drained passively into collecting bottles on the ground by positioning lateral decubitus position using operation table. Manual chest percussion was carried out vigorously during the process of both instillation and drainage2). The initial drainage fluid was milky and opalescent; the procedure was repeated until the effluent was apparently clear (Fig. 4). A total of 10,600 mL of saline was introduced for washing and 8,950 mL was withdrawn. At the end of the procedure, we suctioned out all the remaining fluid using a fibrotic bronchoscope. The whole procedure was completed in 3–4 hours2,4). We had first planned bilateral WLL, of both lungs consecutively; however, we had suspended the left lung lavage due to a hemodynamic instability. Although heart rate and blood pressure were stable, his oxygen saturation decreased down to 68%. The lavage fluid obtained during the first cycle was milky in color. red blood cell was 1,200/µL and white blood cell was 4,690/µL with segmented neutrophil 65%, lymphocyte 10%, monohistiocyte 23%. Cultures of the lavage fluid revealed no growth bacteria and were negative for Acid-Fast Bacillus, Pneumocystis carinii pneumonia, fungi, and Cytomegalovirus culture, culture, respiratory viral study. A similar procedure was done on the left lung 3 days later. A total of 9,935 mL and 8,560 mL were introduced and withdrawn, respectively. He was observed in the ICU for approximately 24 hours thereafter. He was transferred to a ward with no complication.
After the left lung lavage, 3 days later from the initial procedure, his transcutaneous oxygen saturation was more than 95% on room air. Although the patient's pulmonary function did not show significant improvement, he had no problem with breathing on exertion and climbing stairs. On the first follow-up, after 10 days, he was still comfortable, and free from breathing difficulty. His chest X-ray showed markedly cleared lung fields (Fig. 1B). The pulmonary function test, however, demonstrated only limited improvement: there was a restrictive pattern with an FVC of 1.14 L (43% of predicted value), an FEV1 of 1.05 L (44% of predicted value), and a ratio of FEV1/FVC of 92%.
By 4 months after WLL, his pre-WLL symptoms had returned. Chest computed tomography indicated aggravated PAP, and he therefore underwent a second WLL using the same method. Unfortunately, the second WLL did not result in the same level of clinical improvement as the first. Currently, he still has mild dyspnea on exertion, but has no symptoms of vomiting or nausea. We are considering the third WLL with an adjunctive treatment such as the granulocyte-macrophage colony stimulating factor (GM-CSF) or rituximab therapy.
Discussion
PAP, a chronic condition with no cure currently, was first identified in 19581). If left untreated, PAP ultimately upsets pulmonary homeostasis and adversely affects ventilation and perfusion. PAP is characterized by an abundance of surfactant-derived components that accumulate in the lungs because of altered degradation or dysfunction of the surfactant.
This rare disorder is classified as congenital, primary or secondary. Congenital PAP is a genetic condition that results in deficiency of surfactant protein B or abnormality in the beta-chain receptor of GM-CSF. Primary PAP occurs when GM-CSF neutralizing autoantibodies prevent GM-CSF from binding to alveolar macrophage receptors, and accounts for more than 90% of all PAP cases. Secondary PAP can be associated with infection, such as Pneumocystis jirovecii and tuberculosis, systemic inflammatory disorder, such as rheumatoid arthritis and SLE, cancer, immunodeficiency or toxic exposure to silica, aluminum dust, titanium oxide, or other materials. We could not classify this case clearly, as we did not evaluate genetic abnormalities related to surfactant protein B or GM-CSF receptors, nor did we measure GM-CSF auto-antibodies as the necessary test reagents were commercially unavailable in Korea. We suspected that this was not a secondary PAP case, as he had no signs of systemic inflammatory diseases or a history of exposure to noxious materials. Current literature indicates that the frequency of allelic mutations that would result in genetically inherited PAP is rare, given that 90% of PAP cases are classified as autoimmune2). Therefore, the present case is most likely an autoimmune-related primary PAP.
In children, the PAP diagnosis can be easily delayed or missed5). Typical adult patients present with symptoms of progressive respiratory failure and hypoxemia. Children, on the other hand, may present with nonspecific symptoms, such as failure to thrive, vomiting, diarrhea, and dyspnea on exertion2). Moreover, the dyspnea associated with PAP can have an insidious onset, often with no evidence of a respiratory compromise, as is characteristic of the disease in adults. Finally, it is extremely rare in children, and physicians are not likely to suspect this grave condition.
The gold standard for PAP diagnosis is histopathological confirmation, but a CT scan is also useful in a suspected diagnosis. CT findings in adults with PAP have been explored in detail, and include a crazy-paving pattern, which refers to thickened interlobular septa and intralobular lines superimposed on a background of ground glass opacities, often with lobular or geographic sparing6). These findings were identified in our patients and finally confirmed in biopsy. Most commonly, bronchoscopy with BAL and transbronchial biopsy can be performed to confirm the diagnosis of PAP and rule out other conditions. But sampling error may occur due to the patchy nature of the disease. If the diagnosis is not proven by transbronchial biopsy, open lung biopsy or video-assisted thoracoscopic surgery (VATS) is the final step in the diagnostic workup. In our case, we first conducted VATS to distinguish other mimicking conditions. Other diagnostic methods include detecting GM-CSF auto-antibodies and surfactant protein deficiency in serum and bronchoalveolar lavage fluid (BALF).
The literature describes a few therapeutic approaches for the treatment of patients with PAP5). To date, WLL remains the treatment of choice for PAP, irrespective of the cause. It was first described in the early 1960 and the success of WLL was confirmed in adults and adolescents, with response rate of 60%–84%3,7,8). However, for infants and children, information about its efficacy is limited, because the procedure is difficult to perform and is not well tolerated by children. Approximately 15% of patients will suffer a relapse, requiring repeat procedures4,9). In our patient, the vomiting and dyspnea on exertion improved dramatically after the first WLL, although his symptoms gradually worsened there after, and he finally underwent a second WLL 4 months after the first lavage. Despite the second WLL, there was no greater response than after the first lavage. He is currently under observation only, as his symptoms are still tolerable.
Treatments other than WLL are needed if the patient's symptoms were to become further aggravated. GM-CSF supplementation is the first alternative, considering the higher likelihood of an autoimmune cause. After anti–GM-CSF antibodies were discovered in the serum or BALF of patients with primary PAP and their role in PAP pathogenesis were discovered, the possibility of exogenous administration of this cytokine was proposed as a potential treatment10). Both subcutaneous and inhaled GM-CSF therapies were proven to be effective in a number of studies11,12). Recently, rituximab was suggested as another alternative. Rituximab is a monoclonal antibody that targets the CD20 antigen receptor on the surface of the B cells13). It causes a rapid reduction of B cells and acts via complementmediated cytotoxicity, antibody-dependent cell-mediated cytotoxicity, or increased antibody-mediated apoptosis. As primary PAP is considered to be an autoimmune disease involving anti–GM-CSF antibodies, the reduction of B-cell and anti-GM-CSF levels by means of rituximab may be an effective treatment option10,14). However, there have been only few cases where rituximab has been used in children.
If patient do not show any effect despite repeat WLL, GM-CSF, and rituximab treatments, lung transplantation should be considered as a last therapeutic option3). However, lung transplantation is not only dangerous, but there are concerns that the condition may recur if the diagnosis is unclear.
In conclusion, we here report a pediatric PAP case that was confirmed histopathologically, who was treated with a standard WLL procedure, and presented a grave clinical course that required a second WLL procedure due to relapse. This case differs from a previously reported Korean case, which was very mild and self-limited and did not require any intervention15). PAP is not always mild in children and that physicians should keep the possibility of this diagnosis in mind when encountering a child with dyspnea on exertion.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


