


Introduction
3-Methylcrotonyl-coenzyme A carboxylase (3-MCC) deficiency is an autosomal recessive disorder of leucine metabolism1). It is detected in 1 out of 36,000 births, globally2,3). With the expansion of newborn screening programs 3-MCC deficiency has become one of the most commonly diagnosed inborn error metabolisms. This has allowed the identification of asymptomatic mothers, who previously would have been undiagnosed for the disease4). If babies are higher than normal levels of 3-hydroxyisovalerylcarnitine (C5-OH) that is the diagnostic marker for 3-MCC deficiency in normal infants of mothers with 3-MCC deficiency, has been designated maternal 3-MCC deficiency5). We have reported a case of maternal 3-MCC deficiency in which both her infants were noted to have elevated C5-OH on newborn screening tests, and with normal results on metabolic work up including urine organic acid analysis. We confirmed the diagnosis of maternal 3-MCC deficiency by blood spot and breast milk spot test by liquid chromatography-tandem mass spectrometry (LC-MS/MS), urine organic acid analysis and molecular genetic analysis.
Case report
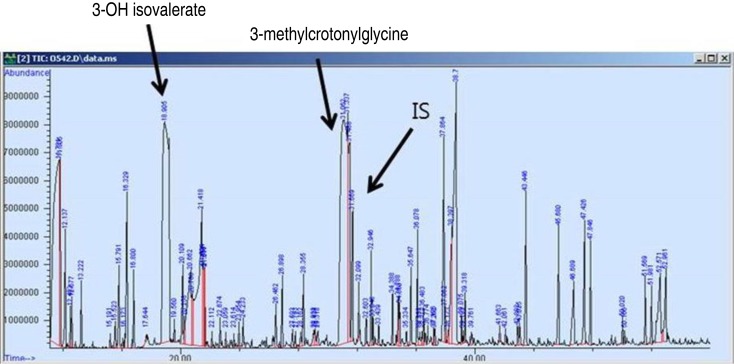
A 15-day-old male with breastfeeding was transferred to the outpatient department due to increased C5-OH detected by LC-MS/MS. His C5-OH levels had been 6.96 µmol/L (reference range, <0.7 µmol/L) at 3 days old, and were 6.02 at 15 days old. His medical history included a normal vaginal delivery at 39 weeks, with a birth weight of 3.120 g. Family history revealed an elder sister who had had increased C5-OH levels of 4.32 µmol/L (reference range, <0.65 µmol/L) at 3 days and 3.67 µmol/L at 7 days old, with nonspecific results on urine organic acid and serum amino acids analysis. A metabolic work up, including a blood spot test by LC-MS/MS, urine organic and serum amino acids analysis, revealed nonspecific results in the 15-day-old male infant. Also, his elder sister at 24 months revealed normal result of C5-OH levels by LC-MS/MS. Maternal 3-MCC deficiency was suspected, and a full metabolic work up was evaluated. The results revealed that maternal serum ammonia, uric acid, lactate and pyruvic acid were within the normal ranges, but the total carnitine, free carnitine and acylcarnitine levels were at 7.96 µmol/L (reference range, 28.00–84.00 µmol/L), 4.14 µmol/L (reference range, 24.00–66.00 µmol/L) and 3.80 µmol/L (reference range, 4.00–32.00 µmol/L), respectively. High C5-OH levels were observed on the maternal dried blood spot by LC-MS/MS, and high levels of excreted 3-hydroxyisovaleric acid (147.9 mmol/mol Cr; reference range, 6.9–25.0) and 3-methylcrotonylglycine (209.4 mmol/mol Cr; reference range, not detectable) were found by the urine organic analysis (Fig. 1).
The C5-OH levels in a dried breast milk spot were especially analyzed by LC-MS/MS. For analysis, dried breast milk spot samples were collected from the infant's mother, and 10 age-matched control mothers. The breast milk spots were prepared as follows: 4 spots were made (volume, 0.05 mL/spot) by dripping (diameter, 1.3 cm/spot), and were dried for 3 hours at room temperature in light protected conditions; breast milks spots were punched out into a 96-well plate, and were quantitated by LC-MS/MS with an API-2000 connected to an Agilent 1100 LC system (AB SCIEX, Toronto, Canada). The results revealed that the maternal breast milk spot contained 27.8 fold higher C5-OH levels than the mean control level (Table 1).
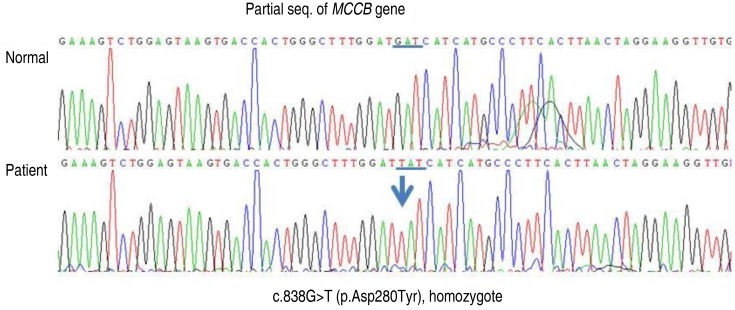
The MCCA and MCCB genes were sequenced for confirmation of mutation. For sequencing, the coding regions of the MCCA and MCCB genes were first amplified by polymerase chain reaction (PCR) from the maternal genomic DNA. The PCR products were the sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied biosystems, Foster City, CA, USA), according to the manufacturer's instructions, and analysis was performed using the ABI3130x1 Genetic Analyzer (Applied biosystems). A c.838G>T (p.Asp280Tyr) homozygous mutation within exon 9 of the MCCB gene was found by the gene mutation analysis (Fig. 2). No mutation within the MCCA gene was observed. Therefore, asymptomatic maternal 3-MCC deficiency was confirmed. The patient is currently receiving carnitine supplemental treatment.
Discussion
3-MCC deficiency is an autosomal recessive disorder of leucine metabolism. It is caused by mutation in the MCCA or MCCB gene coding for α and β subunit, respectively. The phenotype is highly variable ranging from asymptomatic adults to acute neonatal onset with fatal outcome. Grunert et al.1) reported that of the 53 cases identified by newborn screening, 5 asymptomatic patients presented with acute metabolic decompensations later. However, neither the genotype nor the biochemical phenotype was helpful in predicting the clinical course. The most common laboratory findings were ketoacidosis and hypoglycemia. The clinical symptoms were variable such as vomiting, encephalopathy with impaired consciousness, seizure, metabolic stroke, hemiparesis, cerebral edema, muscular hypotonia, muscle weakness, muscle pain, failure to thrive, mental retardation, attention deficit hyperactivity disorders, Reye syndrome and Autism Spectrum disorders1,6). Maternal 3-MCC deficiency may be found by screening for positive normal neonates who have abnormal C5-OH levels. Persistent elevation in C5-OH in the non 3-MCC deficiency neonate could result from placental transfer of C5-OH, C5-OH transfer via the breast milk, and additionally, due to inefficient clearance mechanisms for the metabolite, early in postnatal development2,4,5,7). According to a national survey of extended newborn screening by LC-MS/MS in Taiwan, the overall incidence of inborn error metabolism was approximately 1 per 6,219. The number of cases screened for C5-OH was 592,717, and of these, 4 cases of maternal 3-MCC deficiency were detected8). A single case of maternal 3-MCC deficiency was found among 12,952 cord blood samples in the north of England9). Therefore, infants who test positive on screening and their mother are recommended to have further metabolic analysis such as urine organic acid, plasma acylcarnitine profile and plasma carnitine analysis. In the event that the maternal and the formula-fed infant metabolite levels are both abnormal, the metabolic work up should be repeated in the infant after one month to verify after one month to verify that the infant's levels are normalizing due to clearance of prenatally transferred metabolites; and metabolic laboratory work up should be repeated again in the infant after weaning, because maternal metabolites may be transferred to the infant via breast milk3). One study revealed higher levels of C5-OH level on breast milk samples that was diluted five times with methanol prior to sample processing and quantification than blood reference range2).
We have reported a case of suspected maternal 3-MCC deficiency because of elevated C5-OH levels in two of her infants, by newborn screening with LC-MS/MS and with normal urine organic analysis. We confirmed maternal 3-MCC deficiency by serum and on breast milk spot analysis by LC-MS/MS, urine organic analysis and gene mutation tests. Importantly, we confirmed marked higher levels of C5-OH on breast milk spot by LC-MS/MS, in the case of maternal 3-MCC deficiency vs. controls.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


