




Introduction
Pompe disease (PD), also known as glycogen storage disease type II (GSD II), is caused by a deficiency of lysosomal acid alpha-glucosidase encoded by GAA [1]. This deficiency results in glycogen accumulation in various body systems such as the central nervous system, though it occurs most primarily in the muscle tissue [2]. PD presents as a broad spectrum of clinical phenotypes and can be divided into 2 main groups by age of onset and progression of symptoms: infantile-onset PD (IOPD) and late-onset PD (LOPD). The prevalence of PD ranges from 1:14,000 to 1:300,000 depending on geographic region and ethnicity [3]. IOPD has an apparently higher incidence among African-Americans and Chinese, while LOPD appears to have a higher incidence in the Netherlands [4].
IOPD is the result of a near-complete deficiency of GAA, with an estimated frequency of 1:138,000 [5]. IOPD is characterized by hypertrophic cardiomyopathy (HCMP) in 88% to 100% of patients [6,7]. Symptoms develop at a median age of 18 months if left untreated and include cardiomegaly (92%ŌĆō96%), congestive heart failure (50%), respiratory distress (78%), muscle weakness (63%ŌĆō96%), and feeding difficulty (57%) [6]. The disease can be rapidly progressive. Patients with IOPD who receive intervention and survive infancy have a high prevalence of weakness involving the oropharyngeal muscles, which can lead to difficulty with swallowing and speech [8]. If left untreated, patients typically die within one year [9,10].
LOPD is the result of a partial deficiency of GAA. The estimated prevalence is 1:57,000 [11]. Symptoms usually begin in the first decade of childhood or adulthood. In both children and adults, cardiac involvement is very rare, and symptoms are dominated by significant involvement of the skeletal muscles [4]. Patients with LOPD are characterized by limb girdle weakness; in addition, nonspecific clinical manifestations, including feeding intolerance, recurrent respiratory infection, skeletal deformity, consonant articulation disorder, and persistent diarrhea, should prompt the consideration of PD. The heart is usually not involved. In comparison with IOPD, LOPD is considered a ŌĆ£milderŌĆØ phenotype [12], likely due to higher residual enzyme activity, but there is still significant impact on morbidity, quality of life (QoL), and life expectancy [13].
Enzyme replacement therapy (ERT) for PD with recombinant human acid alpha-glucosidase was approved in both Europe and the United States in 2006. ERT has shown a positive impact on survival, motor development, and HCMP in patients with IOPD [5,14] and has prompted improvement of motor function and/or mobility and pulmonary function in adults with LOPD [2,15]. However, there are limited data on the effect of ERT in children with IOPD [16]. Patients with IOPD usually show rapid progression of generalized muscle weakness, HCMP, and death from cardiorespiratory failure or respiratory infection in the first 1 to 2 years of life [17]. Therefore, early diagnosis and application of ERT are important in patients with IOPD to increase the potential for survival.
This study comprehensively examined the clinical features, genotypes, and long-term outcomes in patients with PD following ERT.
Materials and methods
1. Patients
Five Korean patients (2 males and 3 females) diagnosed with PD between 2002 and 2013 at Samsung Medical Center in Seoul, Republic of Korea, were included in this study. All patients had been previously diagnosed by measurement of acid alpha-glucosidase activity in peripheral blood leukocytes or skeletal muscle biopsy specimens and by mutation analysis.
2. Muscle biopsy
Muscle biopsy specimens measuring approximately 1.5 cm├Ś0.5 cm were dissected through the skin with minimum trauma along the long axis of the fibers. Specimens were held at -140┬░C for 10 to 30 seconds. Specimens were frozen in liquid nitrogen, and fresh-frozen cryostat sections were prepared with hematoxylin and eosin, modified Gomori trichrome, oil red O, periodic acid-Schiff (PAS), cresyl violet, and Sirius Red staining. Histochemical reactions were examined according to standard procedures [18].
3. Molecular analysis
Genomic DNA was isolated from peripheral blood leukocytes according to standard procedures [19]. Polymerase chain reaction was performed to amplify the entire coding region of exons 2 to 20 and the surrounding intronic sequences of the acid alpha-glucosidase gene, as described previously by Ko et al. [20]
4. Clinical evaluation
Conventional doppler and 2-dimensional M-mode echocardiography were performed. According to the pediatric cardiomyopathy registry criteria, HCMP is defined as z score >2 of LV wall thickness. pulmonary function testing (PFT) (in patients [P] 2 and 4), abdominal ultrasonography, skeletal survey, modified barium swallow testing, speech-language evaluation, Denver Developmental Screening Test, Korean-Wechsler Intelligence Scale For Children-Fourth Edition (K-WISC-IV), and Short form 36 (SF-36v2) Health Survey were performed.
5. Alpha-glucosidase IgG antibody measurement
Alpha-glucosidase immunoglobulin G (IgG) antibody was measured with an enzyme-linked immunosorbent assay (ELISA). Antibody titer was the reciprocal of the highest sample dilution in the antibody that was detected by the ELISA.
6. Manual muscle testing
Manual muscle testing (MMT) is a highly reliable method for assessing strength and was used in all patients. During the test, the examiner stood to the side being evaluated, with the patient sitting upright and positioned to allow full movement of the joint against gravity. Grade 3 indicates no resistance, grade 4 indicates moderate resistance, and grade 5 indicates maximum resistance.
7. Six-minute walk test
The 6-minute walk test (6MWT) is practical and simple and requires a 100-foot hallway but no exercise equipment or advanced technician training. This test measures the distance that a patient can quickly walk along a flat, hard surface (e.g., the hallway) in six minutes. Most patients do not reach their maximum exercise capacity during the 6MWT. However, because most activities of daily living are performed at submaximal levels of exertion, the 6MWT may better reflect the functional exercise level for daily physical activities [21]. The test was performed on P2 and P4, as both could walk independently.
Results
1. Clinical characteristics of patients with PD at diagnosis
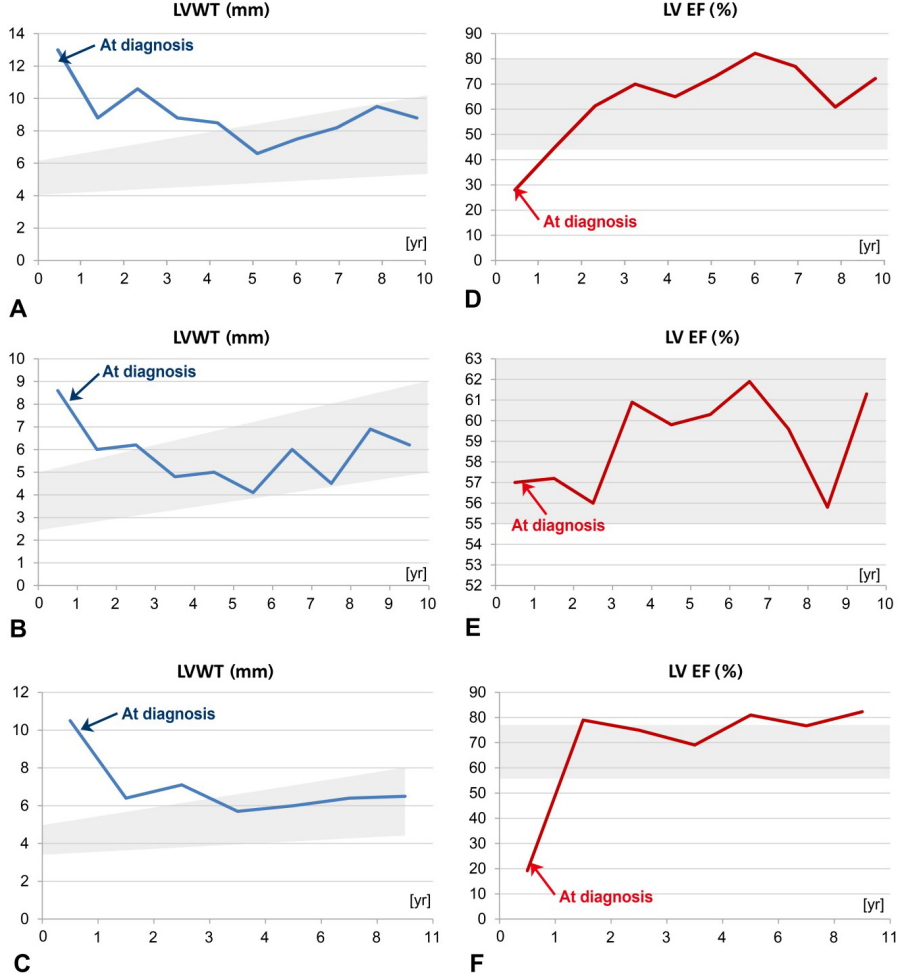
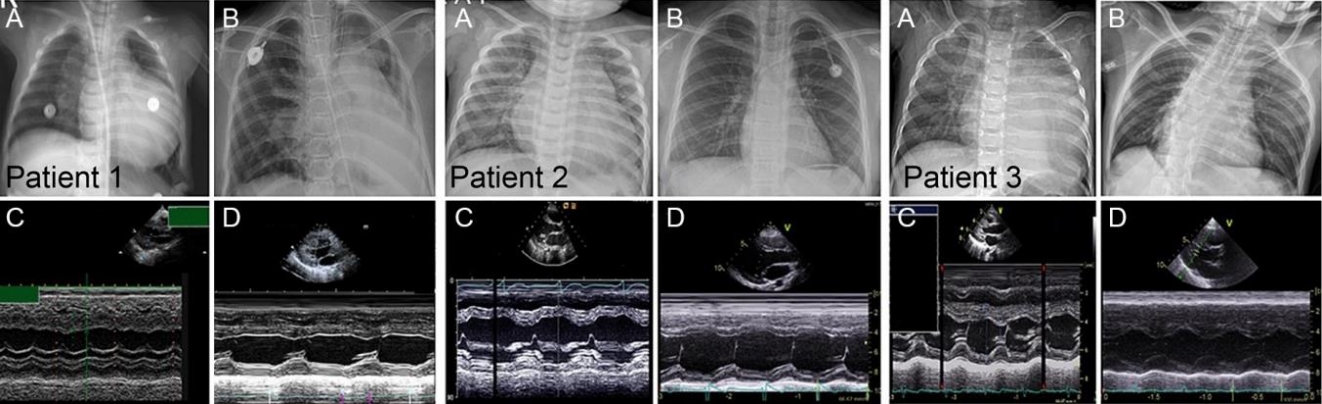
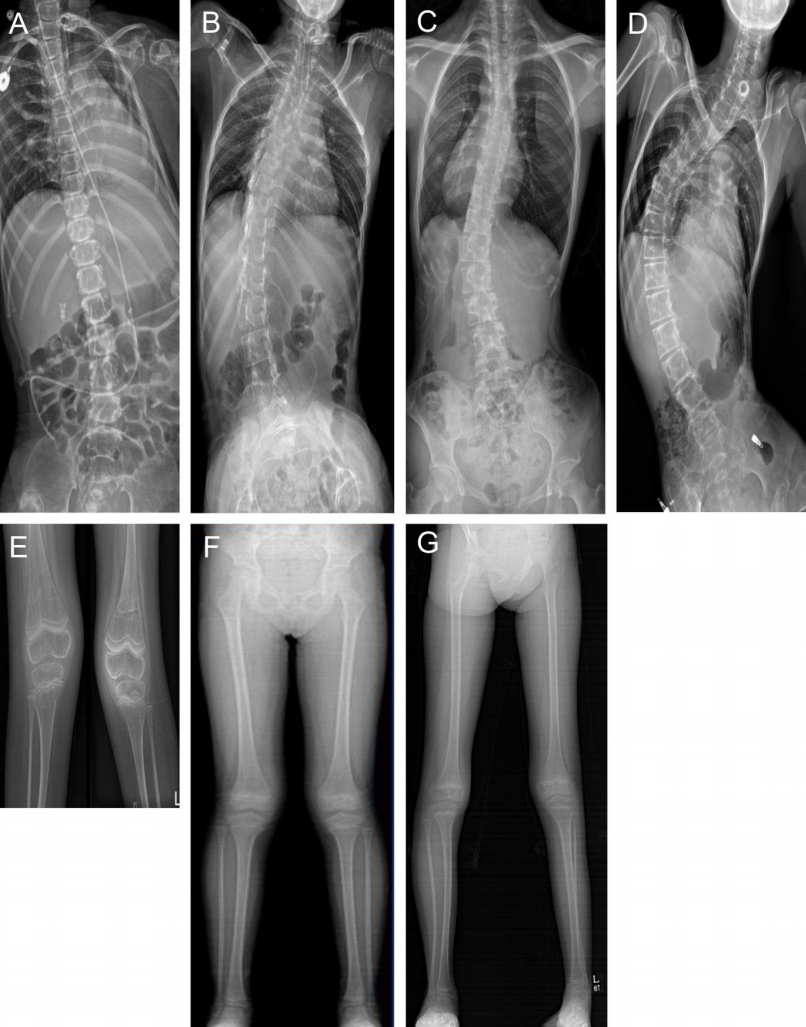
Five patients (2 males and 3 females) with PD were included in this study (Table 1). Three patients (P1, P2, and P3) had IOPD, while 2 (P4 and P5) had LOPD. Initial IOPD symptoms appeared at the age of 1 month in P1 and at the age of 5 months in P2 and P3. Initial LOPD symptoms appeared at the age of 5 years in P4 and at 1 year in P5. Common initial symptoms included hypotonia, cyanosis, and tachycardia in patients with IOPD and limb girdle weakness in patients with LOPD. All patients with IOPD showed HCMP at the time of diagnosis. P2, P3, and P5 were suspected to have PD after histologic examination of skeletal muscle due to hypotonia or muscle weakness (Figs. 1, 2). All diagnoses were confirmed based on acid alpha-glucosidase activity and genetic analysis (Table 2). Intravenous recombinant human alglucosidase alfa (Myozyme, Sanofi, Paris, France) has been available in Korea since 2007. Age at the start of ERT was 5 months in P1, 1 year in P2 and P3, 17 years 6 months in P4, and 6 years 2 months in P5.
2. Enzyme activity and molecular analysis
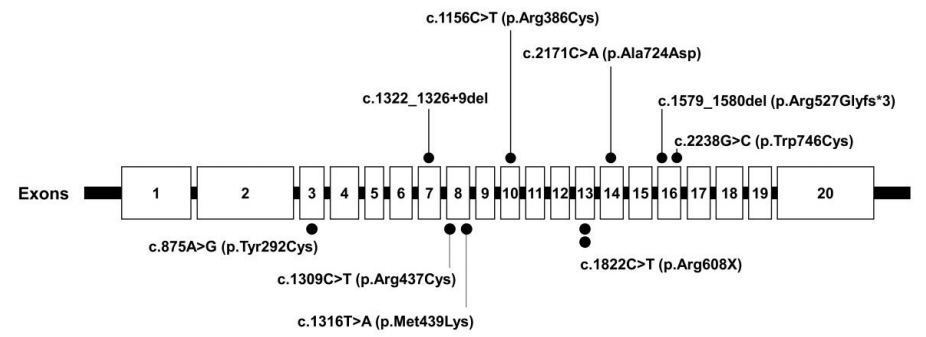
The acid alpha-glucosidase activity in all patients with PD had remarkably decreased below the normal range (Table 2). Sequencing of GAA revealed 9 previously reported pathogenic variants, including 2 nonsense and 5 missense mutations as well as 2 deletions (Fig. 3, Table 2). P1 and P5 had c.1822C>T (p.Arg608X), while P4 carried c.2238G>C (p.Trp746Cys), the most common LOPD mutation in Chinese patients [22].
3. Pathologic findings
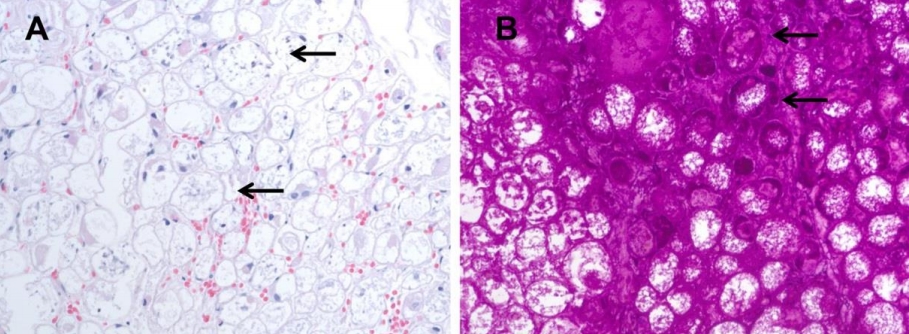
All patients with IOPD had undergone muscle biopsy on suspicion of neuromuscular disease with hypotonia or muscle weakness before nine months of age. Histopathologic examination confirmed the presence of vacuolar changes in myofibers (glycogen-loaded lysosomes) that stained PAS-positive, and further testing for acid alpha-glucosidase was performed. These findings were present in the skeletal muscle of P1 (Fig. 4), P2, P3, and P5 and in the liver of P4. P4 underwent liver biopsy at the age of 7 years due to elevated liver enzymes and hepatomegaly. The liver biopsy showed incomplete PAS(+) fibrous septation on microscopic pathology and excessive accumulation of glycogen particles in the cytoplasmic matrix. Nonlysosomal type was observed on electron microscopy. Therefore, GSD III was suspected. However, the red blood cell glycogen assay was normal. Since the age of 10 years, P4 had had limb girdle weakness and could not walk or run well. At age 17 years, GAA sequencing revealed pathogenic mutations, and a PD diagnosis was made after confirmation of decreased acid alpha-glucosidase activity.
4. Pulmonary function testing
Two patients (P1 and P3) with IOPD and one patient (P5) with LOPD were ventilator-dependent. On PFT, P2 showed normal results. P4 showed a severe restrictive pattern (forced expiratory volume in 1 second/forced vital capacity [FEV1/FVC], 0.81; FVC, 1.17 L; FEV1, 0.95 L). Two patients (P1 and P3) with IOPD experienced recurrent respiratory infections, chronic otitis media, and hearing loss. P1 had respiratory distress from the first month of life and started receiving ventilator support. P3 was doing well without breathing difficulty despite repeated respiratory infections, but suddenly developed pneumonia after 9 years of ERT and subsequently required ventilator support due to respiratory failure. P5, who had LOPD, needed ventilator support from the time of diagnosis.
5. Cardiac evaluation
Three patients with IOPD showed HCMP without outflow tract obstruction (at the age of 2 months in P1 and 9 months in P2 and P3). P3 had moderate mitral regurgitation. These patients showed improvement of ejection fraction and left ventricular posterior wall thickening after ERT (Table 3, Fig. 1, 2). Two patients with LOPD have shown normal echocardiography to date.
8. Swallowing test
P1 underwent gastrostomy and fundoplication at the age of 2 years due to gastroesophageal reflux and swallowing muscle weakness according to a modified barium swallow test. P3 had silent penetration before/during/after swallowing on the test at the age of 11 years and received frequent oral feedings in small amounts. No other patients had swallowing abnormalities (Table 1).
9. Speech-language testing
P2, P3, and P5 showed an articulation disorder in the pronunciation of consonants on speech-language evaluations. The accuracy of consonant pronunciation was expressed as a percentage (Table 1). P1 could not be evaluated due to tracheostomy and severe language developmental delay.
10. Alpha-glucosidase immunoglobulin G antibody measurement
Alpha-glucosidase IgG antibody levels of all patients were measured after 9 years (range, 5ŌĆō11 years) of starting ERT. All 3 patients with IOPD had no alpha-glucosidase IgG antibody, while 2 patients with LOPD showed alpha-glucosidase IgG antibody level of 1,600 U/mL in P4 and 200 U/mL in P5.
11. Developmental delay
Motor, personal-social, and language development was evaluated using the Denver Developmental Screening Test at diagnosis. The motor, personal-social, and language developmental levels in P1 were 5 months, 6 months, and 8 months, respectively, at 13 months. The developmental levels of P3 were 2 months, 5 months, and 8 months at the age of 15 months. P5 had normal language and personal-social development, but motor development was delayed to 20 months at the age of 10 years and eight months. P2 and P4 showed normal development at the time of diagnosis. The recent overall total intelligence of the P2, P4, and P5 measured by K-WISC-IV was 89 score (22.2th percentile), 86 score (18th percentile), 80 score (10th percentile), respectively, with lower scores than the average.
12. Manual muscle testing
P1 and P2 underwent MMT after 9 years of ERT. P1 only showed slight toe movement. P2 was unable to dorsiflex the ankles due to Achilles shortening, but the strength in the other muscles was good. P3 underwent MMT after 4 years and 11 years of ERT. Elbow strength showed no difference, and all other muscles showed reduced strength. P4 underwent MMT after 1 year and 5 years of ERT. In this patient, bilateral upper motor strength was grade 4 and above, but knee flexion/extension remained at grade 3/4; additionally, ankle dorsiflexion/plantarflexion increased from 4/4 to 4/5. P5 underwent MMT after 6 years and 12 years of ERT. Upper/lower motor strength decreased from 3/4 to 2/3 (Table 4).
13. Sort Form 36 Health Survey
The SF-36v2 Health Survey uses 36 questions to measure functional status and was performed at the ages of 10 years 4 months in P1, 10 years 8 months in P2, 12 years 2 months in P3, 22 years 8 months in P4, and 18 years 3 months in P5 (Table 5). The physical component score (PCS) in P1, P3, and P5, who were wheelchairdependent, was less than 20 points, while the PCS in P2 and P4, who showed no progression of muscle weakness on MMT, was 70 points or more after ERT. In P2 with IOPD, ERT began at the age of 12 months, and this patient showed a higher total score for the survey, with a mental component score of 85.3125 and a PCS of 72.5 (Table 5).
14. Side effects of ERT in patients with PD
All patients continued to receive ERT without side effects, such as sweating, headaches, elevated temperature, or hypotension via infusions of recombinant human alglucosidase alfa [23].
Discussion
This study examined clinical and molecular characteristics and the 10-year experience of ERT in 5 patients with IOPD and LOPD in a single center in Korea. Three patients (P1, P2, and P3) were diagnosed with IOPD and experienced initial symptoms by the age of 5 months. These individuals developed HCMP within 1 year after birth, but all have remained alive on ERT. Two patients (P4 and P5) were diagnosed with LOPD, and their initial symptom was limb girdle weakness. The most common initial symptom was cardiomegaly and muscle weakness in IOPD and LOPD, respectively. Cardiomegaly (100% vs. 0%) was found only in patients with IOPD, while hepatomegaly (66.7% vs. 100%) and muscle weakness (66.7% vs. 100%) were observed more frequently in patients with LOPD (Table 1). Respiratory assistance was required in 66.7% of IOPD patients and 50% of LOPD patients, and wheelchairs were required in 60% of PD patients.
The GAA gene contains 20 exons and is highly polymorphic, with a large number of neutral variations. Thus far, 558 sequence variants of the GAA gene have been published in the Pompe Disease Mutation Database. The finding of some frequently occurring GAA mutations in certain populations points to single mutational events in those populations. The best-documented example is the African-American mutations, c.2560C>T(p.Arg854X) [24]. A second well-known example of a founder mutation is c.1935C>A (p.ASP645Glu). [25]. c.1726G>A (p.Gly576Ser) and c.2065G>A (p.Glu689Lys) are more frequent among Asian populations [26,27]. These 2 sequence variants are most often found together on the same allele, and an estimated 3.3% to 3.9% of people in Asian populations are homozygous for both variants [28]. c.2481+102_2646 + 31del (deletion of exon [18] is common in some subsets of the Caucasian population [29]. Some mutations appear with considerable frequency in particular ethnic groups. The c.546G>T (p.Thr182Phe) (22.9%, 16 of 70) was found to be the most common mutation, followed by c.1857C>G (p.Ala619Arg) (14.3%, 10 of 70), in Japanese patients with PD [30]. The c.1935C>A (p.Asp645Glu) mutation accounted for 80% (8 of 10 allele) of the GAA mutations found in the Thai population [31]. In Spanish patients with PD, the 2 most common GAA mutations, at frequencies of 25% and 13.6% respectively, were 11/44 allele (c.-32-13T>G,p.Leu705Pro) and 6/44 allele (c.1076-1G>C) [32]. Additionally, the c.1857C>G (p.Ala619Arg) (23.3%, 7 of 30 allele) and c.1316T>A (p.Met439Lys) (13.3%, 4 of 30 allele) mutations were revealed as the most common in Korean PD patients [33]. In the present study, c.1316T>A (p.Met439Lys) was detected in P2, who had IOPD. All PD patients had compound heterozygous mutations. Five missense mutations (c.875A>G (p.Tyr292Cys), c.1316G>A (p.Met439Lys), c.2171C>A (p.Ala724Asp), c.1156C>T (p.Arg386Cys), and Clinical and GAA gene mutation analysis in mainland Chinese patients (p.Arg437Cys)); 2 nonsense mutations (c.1822C>T (p.Arg608X) and c.2238G>C (p.Trp746Cys)); and 2 deletion mutations (c.1579_1580del (p.Arg527Glyfs*3) and c.1322_1326+9del) were identified. No hotspot was found, and c.1822C>T (p.Arg608X) was found in 2 patients, specifically P1 (IOPD) and P5 (LOPD). P4 carried 2238G>C (p.Trp746Cys), the most common LOPD mutation found in Chinese patients [22]. c.1822C>T (p.Arg608X) was identified as the second-most common mutation considering only IOPD in Japanese patients [30]. c.-32-13T>C (p.Leu705Pro) was the most common mutation first signs of the disease present before the age of 1 [24].
ERT is not curative but does modify the disease course in patients with IOPD and LOPD. Patients with IOPD usually show rapid progression of the disease and die in the first 1 to 2 years of life without ERT. In this study, ERT was effective for improving survival, likely due to improvement of cardiomegaly and the thickness of the left ventricle in patients with IOPD (Table 3). Our patients with IOPD showed a stable disease course during a median of 10 years of follow-up. Early identification of IOPD is very important, as ERT increases lifespan and treats HCMP, hepatomegaly, and elevated liver transaminases despite persistent respiratory distress. Also, ERT was effective for treating a hepatomegaly, elevated liver transaminase level, and to increase partially (only P4) in strength of muscle in LOPD.
According to the existing literature, some patients develop infusion-associated reactions such as sweating, headaches, elevated temperature, or hypotension during infusions of recombinant human alglucosidase alfa [23]. However, in this study, all patients continued to receive ERT without side effects.
According to the existing literature, patients with PD have significantly lower QoL values in physical functioning, general health, vitality, and social functioning scales [34]. This study illustrated that, in patients (P2 and P4) with little muscle weakness and a lack of reliance on a wheelchair, QoL assessment revealed higher physical functioning and mental health component scores than in other patients. Interestingly, P5 had worsened muscle weakness and was dependent on a wheelchair, but he had the highest mental component score, even though his physical functioning score was low.
In PD, development of high titers of anti-GAA antibodies was primarily affected by cross-reactive immunologic material (CRIM) status. Therefore, CRIM status is an important predictor of response to ERT. In our study, none of the patients could be tested for CRIM status prior to ERT, but neutralizing antibodies that can influence the bioavailability of recombinant enzyme were measured. All 3 patients with IOPD had no alpha-glucosidase IgG antibody, while two patients with LOPD showed positive alpha-glucosidase IgG antibody level. Alpha-glucosidase IgG has been demonstrated to have a negative impact on ERT outcomes in one study [5]. Neutralizing antibodies developed in all treated patients, whether IOPD or LOPD [5]. IgG antibody titer showed peak level 12 weeks after ERT, and declined slowly thereafter. Higher infusion dose (mg/kg) was associated with higher antibody production [5]. However, IgG antibody titers did not correlate with treatment efficacy in other study. We will follow up alpha-glucosidase IgG antibody titers for all patients in this study.
In the present study, respiratory assistance was required in 60% of PD patients. As impaired respiratory function is associated with high morbidity and impaired QoL, better treatments are needed. Alglucosidase alfa (Myozyme) is a recombinant human alphaglucosidase used for treatment of IOPD and LOPD. Several clinical trials and investigations for treatment of PD have been performed. To increase penetration into muscle cells and clear a greater substrate, new drugs are being developed. Neo-GAA (next-generation ERT) phase III trials are ongoing in naïve patients with LOPD in the United States and European Union. VAL-1221 (Valerion Therapeutics, Concord, MA, USA) is a therapeutic agent being considered in phase II clinical trials ongoing in patients with LOPD in the United Kingdom and the United States and is designed to deliver GAA to the cytoplasm and autophagic vacuoles as well as lysosomes. Duvoglustat coformulated with a proprietary ERT using the CHART Platform (Amicus Therapeutics, Cranbury, NJ, USA) is a nextgeneration small-molecule pharmacological chaperone that is part of a phase II trial ongoing in LOPD patients in Australia, Germany, the Netherlands, the United Kingdom, and the United States. Gene therapy using a recombinant adeno-associated viral vector (Audentes Therapeutics, San Francisco, CA, USA) is part of a phase I trial ongoing in patients with LOPD in the European Union and the United States.
The present studyŌĆÖs limitations include the small number of patients and a lack of serial clinical parameters. However, in this study, the relatively long-term clinical course of IOPD was well-described, and these results indicate the effectiveness of ERT for IOPD. Therefore, patients with IOPD should be considered potential candidates for clinical trials of new drugs in the future.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


