




Introduction
There are 2 important components of hemostasis: primary (platelet response to vessel injury and platelet plug formation) and secondary (generation of fibrin deposition by the coagulation cascade) [1]. Platelets play a key role in the achievement of adequate primary hemostasis through 4 fundamental mechanisms: adhesion, aggregation, secretion, and procoagulant activity [2,3]. Normal platelet adhesion requires the presence of von Willebrand factor (vWF), glycoprotein (GP) receptors on platelets, and the adequate release of platelet granular contents such as adenosine diphosphate (ADP), serotonin, and thromboxane A2 [1]. Thus, familial inherited thrombocytopenia, platelet function disorders, von Willebrand’s disease (vWD), or immune thrombocytopenia show similar primary hemostatic problems [4].
However, the diagnostic confirmation and differential diagnosis of IPDs are complicated, time-consuming, and expensive; thus, many patients with IPD are not appropriately diagnosed [4-6]. In most cases, individual megakaryopoiesis, platelet formation, and platelet function are genetically controlled; more than 50 genes associated with IPDs have been identified in recent studies by genome-wide association study and next-generation sequencing [7]. These genes can be classified according to the specific pathway that is disrupted in platelet formation, e.g., the thrombopoietin (THPO)/THPO receptor (MPL) signaling pathway, transcriptional regulation, granule formation/trafficking/secretion, cytoskeleton regulation, and transmembrane protein signaling pathway (Fig. 1) [7].
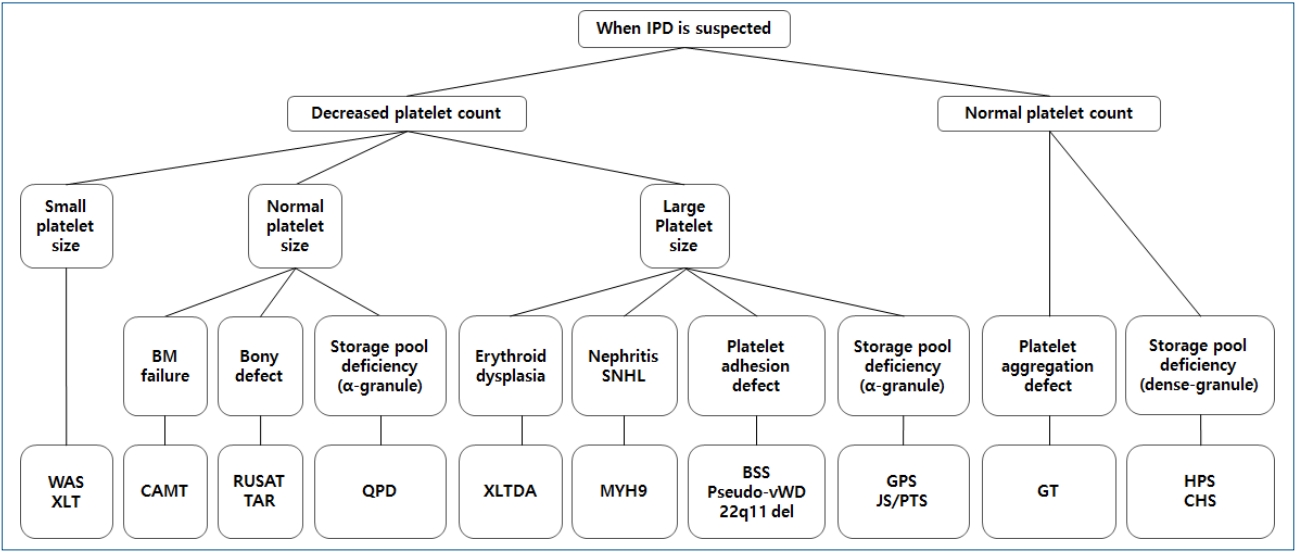
Therefore, in this review, representative IPDs based on genetic classification (Table 1) and the current status of these diseases in Korea (Table 2) will be discussed. In particular, this review focuses on IPDs with thrombocytopenia or thrombocytopathy. The diagnostic sequence and flow in IPD is described in Fig. 2 [4,8].
THPO/MPL signaling pathway defects
1. Congenital amegakaryocytic thrombocytopenia
Congenital amegakaryocytic thrombocytopenia (CAMT) (OMIM #604498) is a rare autosomal recessive IPD with severe thrombocytopenia from infancy due to MPL gene mutation and high levels of serum THPO [9]. The case of a genetically confirmed Korean patient with CAMT harboring a novel mutation in the MPL gene was recently reported [10]. CAMT is characterized by megakaryocytopenia in the bone marrow, thrombocytopenia with normal platelet size, and progressive bone marrow failure to aplastic anemia [9]. Approximately 10%–30% of patients with CAMT have orthopedic/neurological abnormalities or intracranial hemorrhage [6]. The THPO receptor, encoded by the MPL gene, promotes megakaryocyte growth and proliferation and may play a role in maintaining hematopoietic stem cells [9,11]. CAMT with bone marrow failure to aplastic anemia is best managed by a hematopoietic stem cell transplantation (HSCT) [12].
Defects in transcriptional regulation
1. X-linked thrombocytopenia with or without dyserythropoietic anemia
X-linked thrombocytopenia with or without dyserythropoietic anemia (XLTDA) (OMIM #300367) is a rare X-linked recessive IPD with an unknown prevalence; only a few cases have been reported worldwide [13-16]. No Korean cases of XLTDA associated with GATA1 have been reported to date. The GATA1 gene encodes a crucial transcription factor, GATA-binding factor 1, involved in the development of erythrocytes and megakaryocytes in the early stage of hematopoiesis [14]. Depending on the specific GATA1 mutation, macrothrombocytopenia, variable severity of anemia, hemolysis, and hypercellular marrow with erythroid dysplasia can occur [2].
2. Jacobsen syndrome and Paris-Trousseau syndrome
Jacobsen syndrome (OMIM #147791) and Paris-Trousseau syndrome (OMIM #188025) are rare autosomal dominant IPDs associated with the deletion of chromosome 11q [17,18]. A few Korean cases have been reported [19-21]. These diseases share several similar phenotypes (dysmorphic features, intellectual disability) and macrothrombocytopenia with giant α-granules due to the fusion of smaller organelles [17,22]. Chromosome 11q23.3 includes the FLI1 gene, which encodes an essential transcription factor for megakarypoiesis [23]. Hemizygous deletion of the FLI1 gene prevents progenitor cells from undergoing the normal differentiation process, thereby generating a large population of small immature megakaryocytes that undergo lysis [23].
3. Radioulnar synostosis with amegakaryocytic thrombocytopenia
Radioulnar synostosis with amegakaryocytic thrombocytopenia (RUSAT), consisting of RUSAT1 (OMIM #605432) and RUSAT2 (OMIM #616738) types, is a recently identified and introduced autosomal dominant IPD characterized by progressive bone marrow failure and pancytopenia requiring HSCT [24,25]. Only a few families with these conditions have been reported worldwide [2], and no Korean cases have been described in the literature. These syndromes involve amegakaryocytic thrombocytopenia and skeletal defects (limited forearm pronation/supination due to proximal fusion of the radius and ulna) [24,25]. RUSAT1 and RUSAT2 are caused by variants in HOXA11 and MECOM genes, respectively [24,25]. The protein encoded by the HOXA11 gene is a DNA-binding transcription factor that regulates gene expression, morphogenesis, and differentiation [24]. The oncoprotein EVI1, encoded by the MECOM gene, is also a transcriptional regulator involved in hematopoiesis and stem cell self-renewal in humans [26]. In mice, MECOM protein is expressed at high levels in the embryonic limb bud, suggesting that this gene is also involved in limb development [27].
4. Thrombocytopenia-absent radius syndrome
Thrombocytopenia-absent radius (TAR) syndrome (OMIM #274000), also known as chromosome 1q21.1 deletion syndrome, is a rare autosomal recessive IPD with congenital hypomegakaryocytic thrombocytopenia and bilateral radial aplasia [28]. A few Korean cases have been reported to date [29,30]. A mutation in RBM8A gene was recently shown to cause TAR syndrome [31]. RBM8A has several important cellular functions in the production of other proteins by facilitating mRNA transport.31) In TAR syndrome, thrombocytopenia becomes less severe over time; the platelet levels reportedly normalized in some cases [2,6,28]. Some patients also have other congenital anomalies, e.g., lower-limb anomalies, cow’s milk intolerance, renal anomalies, cardiac anomalies, intracranial vascular malformation, facial hemangioma, sensorineural hearing loss, and scoliosis [28]. A skeletal evaluation and bone marrow study are important in thrombocytopenia such as RUSAT or TAR. Two diseases can emphasize that a thorough orthopedic examination is necessary for patients with thrombocytopenia.
Defects in granule formation, trafficking, or secretion
1. Hermansky-Pudlak syndrome (dense granule disorder)
Hermansky-Pudlak syndrome (HPS) is a rare heterogeneous autosomal recessive IPD group with several genetic defects, including mutations in HPS1 (OMIM #203300), AP3B1 (HPS2, OMIM #608233), HPS3 (OMIM #614072), HPS4 (OMIM #614073), HPS5 (OMIM #614074), HPS6 (OMIM #614075), DTNBP1 (HPS7; OMIM #614076), BLOC1S3 (OMIM #614077), and BLOC1S6 (OMIM #614171) [7,32]. HPS is a disease group of defects in lysosomes, melanosomes, and granule biogenesis [33]. These conditions share features such as oculocutaneous albinism and bleeding symptoms due to platelet dysfunction associated with dense granule defects [2,6,32]. Owing to the presence of dense granule defects, HPS platelets result in aggregation defects with the absence of second-wave aggregation in response to adrenaline [2]. Proteins encoded by the HPS gene are involved in intracellular vesicle transport, protein sorting, and vesicle docking/fusion [2,33]. Additional manifestations, including pulmonary fibrosis, granulomatous colitis, neutropenia, or immunodeficiency, can occur according to mutation type [2,6,32]. No Korean cases of HPS have been reported; however, HPS is a common genetic disorder in Puerto Rico, affecting 1 in 800 individuals, with more than 500 reported cases [34].
2. Chediak-Higashi syndrome (dense granule disorder)
Chediak-Higashi syndrome (CHS) (OMIM #214500) is a rare autosomal recessive IPD characterized by ocular cutaneous hypopigmentation, platelet dysfunction, immunodeficiency due to abnormal natural killer cell function, and peripheral neuropathy associated with mutations in the lysosomal trafficking regulator (LYST) gene [35,36]. Thus, CHS disrupts the structure and function of lysosomes and related structures in cells. HPS and CHS share common clinical manifestations (oculocutaneous albinism and platelet dysfunction due to a dense granule defect) but can be differentially diagnosed by the presence of large peroxidase-positive cytoplasmic granules in neutrophils and more severe symptoms at an earlier age in the case of CHS [35]. Few Korean CHS cases have been reported to date [37].
Defects in proplatelet formation and cytoskeleton regulation
1. MYH9-related disorders
MYH9-related disorders (OMIM #155100) are autosomal dominant IPDs including May-Hegglin anomaly (MHA), Fechtner syndrome (FTNS), Sebastian syndrome (SBS), and Epstein syndrome (ES) [38]. All of these syndromes are associated with the MYH9 gene, which encodes nonmuscle myosin heavy chain IIa (NMMHC-IIA), a component of the contractile cytoskeleton in many tissues, including megakaryocytes, platelets, leukocytes, and the kidneys [38,39]. The altered distribution of NMMHC-IIA within platelets can be measured by immunohistochemistry or immunofluorescence [39]. Thrombocytopenia resulting from defective megakaryocytosis and typical cytoplasmic basophilic inclusion bodies of leukocytes in MYH9-related disorders is caused by the aggregation of abnormal NMMHC-IIA [6]. According to several studies, MHA, FTNS, SBS, and ES are not separate entities but a single disorder with a wide spectrum of clinical symptoms, e.g., from mild macrothrombocytopenia to the more severe form with sensory neural hearing loss, renal failure, and cataracts [40-42]. Platelet counts are typically in the range of 20–130×109/L, with elevated mean platelet volume and mild bleeding symptoms [6,42]. To date, several Korean MYH9-related disorders confirmed by genetic testing have been reported [43-48].
2. Wiskott-Aldrich syndrome and X-linked thrombocytopenia
Wiskott-Aldrich syndrome (WAS) (OMIM #301000) is a rare X-linked recessive IPD, with an incidence of approximately 1–4 per 1,000,000 live male births [2,6]. Although the prevalence of most IPDs in Korean populations has not yet been determined, the prevalence of Korean WAS is 0.33 per 1,000,000 individuals based on an epidemiological study performed using data of the national registry for primary immunodeficiencies [49]. The WAS protein (WASP), expressed in hematopoietic cell lineages, has a fundamental role in signal transduction from receptors on the cell surface to the actin cytoskeleton [2,6,50,51]. Thus, aberrant WASP expression results in defective proplatelet formation and diverse clinical manifestations of WAS, e.g., microthrombocytopenia, eczema, frequent infection due to immunodeficiency, and even malignancy [2,6,50,51]. Microthrombocytopenia is the characteristics of WAS, and present at birth and varies between 5 and 50×109/L [2]. Malignancies occur in 13% of patients with WAS at a mean 9.5 years of age [52].
X-linked thrombocytopenia (XLT) (thrombocytopenia 1; OMIM #313900), the milder variant, is characterized by thrombocytopenia with transient eczema and absent or minimal immunodeficiency [51]. WAS gene-associated XLT can be misdiagnosed as chronic ITP because it is milder without frequent infection or eczema [6]. Several cases of Korean patients with WAS or XLT confirmed by genetic testing have been reported to date [53-60]. The results of hematopoietic stem cell transplantation in Korean patients with WAS have also been reported [54,58,61]. XLT is difficult to be differentially diagnosed clinically from XLTDA (OMIM #300367). In the case of XLT, there may be a family history of WAS, which will help with differentiation between the 2 diseases. In addition, platelet size (large in XLTDA, small in XLT) is helpful for differentiating between the 2 diseases.
Transmembrane GP signaling pathway defects
1. Glanzmann thrombasthenia
Glanzmann thrombasthenia (GT) (OMIM #273800) is a rare autosomal recessive IPD with a prevalence of approximately 1 in every 1,000,000 individuals, although a higher prevalence was observed in some populations owing to increased rates of consanguineous marriages [62]. GT typically develops with loss-of-function variants of the ITGB2A or ITGB3 genes encoding GPIIb and GPIIIa, respectively [63]. However, very rare gain-of-function (GOF) variants in the ITGB3 gene have also been reported, resulting in enhanced fibrinogen binding and severe bleeding [64]. To date, several Korean cases of GT have been diagnosed clinically or confirmed by flow cytometry; of them, some were further confirmed by genetic analysis [65]. In patients with GT, there are quantitative and/or qualitative defects in the platelet GPIIb/IIIa complex at binding sites for fibrinogen, vWF, and fibronectin [66,67]. Thus, patients with GT exhibit normal platelet count/morphology but severely diminished platelet aggregation in response to ADP, epinephrine, serotonin, thrombin, and collagen. Platelet function analyser-100 (PFA-100) measurements are significantly abnormal in cases of GT, with prolonged closure times on ADP/collagen and adrenaline/collagen cartridges [2]. Impaired platelet agglutination in patients with GT is corrected by ristocetin, which induces the binding of vWF with GPIb/IX [2]. The application of flow cytometry using antibodies to GPIIb (CD41)/GPIIIa (CD61) is also useful for the diagnosis of GT [2].
2. Bernard-Soulier syndrome
Bernard-Soulier syndrome (BSS) (OMIM #231200) is a rare IPD that is typically transmitted in an autosomal recessive manner; its prevalence is estimated to be less than 1 in 1,000,000 individuals [6]. To date, fewer than 1,000 BSS cases have been reported and studied worldwide [2,6,68]. No Korean BSS cases have been published to date. BSS is characterized by low platelet counts, typically ranging from less than 30 to 200×109/L, and the presence of large platelets (macrothrombocytopenia) in a peripheral blood smear [6,69]. These large platelets are associated with the abnormal development of platelet membranes due to GPIba abnormalities [70]. In patients with BSS, platelets have defective surface expression of GPIb-IX-V, which acts as a vWF receptor on platelets, owing to mutations in the GP1BA, GP1BB, or GP9 genes [2,69,71]. As a result, platelets in patients with BSS cannot adhere to the vascular subendothelium and are unable to be agglutinated by ristocetin [68]. In a study by the International Consortium for BSS, 211 BSS families had mutations in the GP1BA (28%), GP1BB (28%), or GP9 (44%) genes [68]. The closure time in PFA-100 is significantly prolonged in BSS on both cartridges [2]. The detection of defects in the GPIb-IX-V complex by antibodies to GPIb (CD42b) on flow cytometry is also useful for making the diagnosis of BSS [72].
3. Pseudo-vWD
Pseudo-vWD (OMIM #177820), also known as platelet-type vWD or bleeding disorder platelet-type 3, results from GOF variants in the GP1BA gene [73]. Pseudo-vWD is an autosomal dominant genetic disorder of the platelets, and genetic tests of the vWF gene are normal [73]. GOF variants in GP1BA affect the qualitatively altered GPIb receptor, which then shows increased affinity to vWF [74]. Accordingly, large platelet aggregates and high-molecular-weight vWF multimers are removed from circulation, resulting in thrombocytopenia and altered vWF multimers. Ristocetin cofactor activity and altered vWF multimers are similar to characteristics of vWD type 2B, a disorder of GOF variants in the vWF gene resulting in altered function of the A1 domain of vWF [75]. In both cases, increased affinity of the GP1b receptor and vWF A1 domain results in thrombocytopenia and abnormal vWF multimers. Thus, DNA analysis is required for differential diagnosis [7].
4. 22q11 deletion syndromes
The 22q11.21 deletion syndromes, including DiGeorge syndrome (OMIM #188400) and velocardiofacial syndrome (OMIM #192430), may be related to BSS phenotype if the remaining single GPIBB gene contains an independently inherited mutation [76]. These 22q11 deletion syndromes are autosomal dominantly inherited disorders with an estimated incidence of 1 in 4,000 births and are characterized by congenital heart disease, specific facial features, thymic aplasia, frequent infections, cleft palate, hypocalcemia, hypoparathyroidism, learning disabilities, and developmental delay [77]. If patients must undergo heart surgery because of congenital heart disease associated with 22q11.2 deletion syndrome, physicians should be aware of the serious bleeding risk in these patients [78].
Conclusions
In the last few decades, researchers have elucidated many of the etiologies of IPDs. The genes involved in a variety of IPDs have been identified, and molecular characterization of these disorders is underway. This information enables a more accurate understanding of IPDs. To enable more accurate and definitive diagnostics, advanced genetic approaches, including next-generation sequencing, are required for patients with suspected IPD [79,80]. Although the NGS panel for IPDs has not yet been commercialized and popularized in Korea, interest in IPDs is increasing. As described in Table 2, Korean Pediatric Hematology Oncology Group recently attempted the genetic confirmation of IPDs using targeted exome sequencing as a multicenter study. Although this study has only just started, its findings are expected to be useful for future large-scale studies and the establishment of a Korean IPD registry. It also may be useful for the future development of an NGS panel for IPDs in Korea.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


