




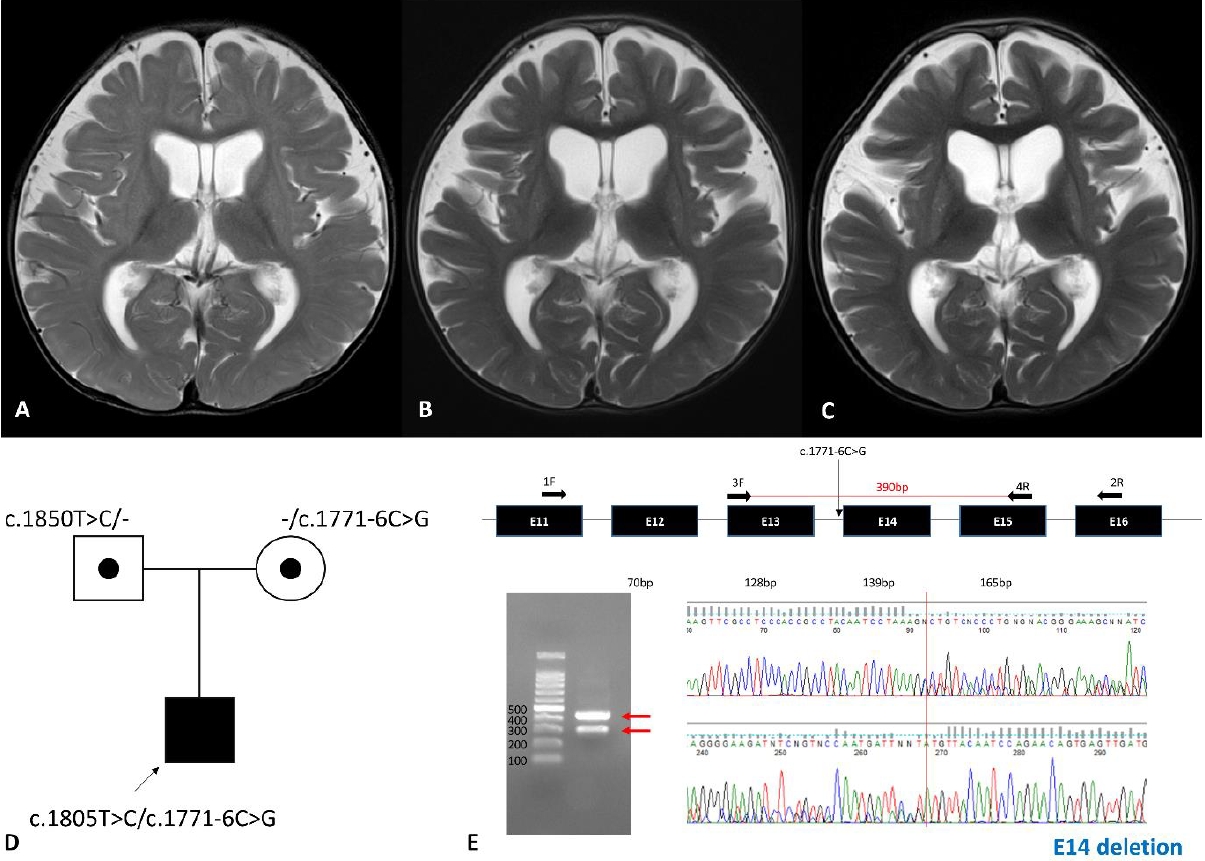
The patient was born from nonconsanguineous and healthy parents at gestational age 40+5 weeks, and his birth weight was 3.1 kg (5th–10th percentile). Fetal growth was retarded during the last month of pregnancy. At 5 months of age, the patient visited the hospital due to poor weight gain. His weight was 5.7 kg (<3rd percentile). He showed progeroid facial appearance with triangular face, droopy skin, maxillary hypoplasia, and sparse hairs. The patient also showed frontal bossing, micrognathia, and lack of subcutaneous fat especially in trunk and buttocks. He was hypotonic and he could not control his head nor roll over his body. Nutritional support using high calorie milk and enteral tube was ineffective to gain weight due to frequent vomiting and aspiration. His anthropometric values reached to 91 cm (<3rd percentile) and 11 kg (<3rd percentile) at the last follow-up of 40 months old. He remains near bed-ridden state at 40 months despite constant physical rehabilitation. His muscle tone has increased with positive upper motor neuron signs and required muscle relaxants with time. He never had social communication skills except brief eye contact. He had pectus excavatum and congenital nystagmus. The ophthalmic examination revealed that he had mild hyperopia and astigmatism. Delayed eruption of upper central incisors, and enamel hypoplasia was observed in dental examination. Laboratory tests including metabolic screening were unremarkable. Brain magnetic resonance imaging (MRI) indicated nonspecific mild brain atrophy at 9 months, but progressed diffuse brain atrophy and T2 high signal intensity of bilateral basal ganglia was observed at 15 and 22 months (Fig. 1A-C). Compound heterozygous variants, c.1771-6C>G and c.1805T>C, in POLR3A were identified by exome sequencing for the patient and his parents (Fig. 1D). We identified the pathogenicity of the variant c.1771-6C>G by real-time polymerase chain reaction followed by Sanger sequencing (Fig. 1E). The variant results in exon 14 deletion.
POLR3A-related disordes
POLR3A (RNA polymerase III subunit A) encodes the largest one of the 17 subunits that constitute Pol III, responsible for transcription of ribosomal 5S RNAs, transfer RNAs, and other small RNAs [1]. POLR3A has been known as the causative gene of hypomyelinating leukodystrophy 7(HLD7, MIM#607694) or Wiedemann-Rautenstrauch syndrome (WDRTS, MIM#264090). HLD7, also known as the 4H (hypomyelination, hypodontia, and hypogonadotropic hypogonadism) leukodystrophy, is an autosomal recessive leukodystrophy mainly presenting with diffuse hypomyelination of white matter and various neurologic symptoms [2,3]. Nonneurologic features such as short stature and ocular abnormalities were also reported in some patients [4]. WDRTS, also known as neonatal progeroid syndrome, mainly displays intrauterine and postnatal growth retardation, a progeroid appearance, hypotonia, generalized lipodystrophy, and dental anomalies [5,6]. Neurologic manifestations such as hypotonia, seizures, and ataxia may also be present in some patients [5-8]. Although their main features are quite characteristic to distinguish each other, HLD7 and WDRTS share some clinical manifestations. Above we reported another atypical case which presented an early-onset lipodystrophy with progeroid facial appearance of WDRTS and devastating neurologic deterioration with atypical brain involvement, which suggested the clinical continuum of POLR3A-related disorder.
Phenotypic variability in POLR3A-related disorder
Over 150 patients with biallelic POLR3A variants have been reported to date (Table 1). HLD7 is the representative and the most frequent disease entity in the POLR3A-related diseases. Most patients with the classic form of HLD7 showed diffuse cerebral hypomyelination. About half of HLD7 patients had significant motor developmental delay but most patients could walk independently. Others achieved normal developmental milestones but usually regressed later. Less than half of HLD7 cases showed short stature. WDRTS cases have distinct clinical features of congenital- or neonatal-onset failure to thrive and lipodystrophy. Motor milestones were delayed in some patients, yet all patients achieved independent walking. Unlike the classic form of HLD7 and WDRTS, atypical form of leukodystrophy or striatal variant cases was recently reported [9-12]. Although most patients with striatal variant showed developmental delay and other neurologic features in common with classic HLD7, their clinical courses seemed to be more severe and rapidly progressive. Their MRI findings were quite different from the HLD7. The focal striatal involvement was representative feature instead of diffuse cerebral hypomyelination. These various disease categories suggested phenotypic variability of POLR3A-related disorder. In this report, we present a boy with severe failure to thrive, progeroid facial features, profound developmental delay with progressive brain involvement, who were confirmed to have biallelic variants of c.1771-6C>G and c.1805T>C in POLR3A. Both his main complaint of severe failure to thrive and examination findings of progeroid facial appearance and lipodystrophy were typical characteristics of WDRTS. The patient, however, also showed profound developmental delay and progressive striatal involvement.
Discussion of this case: WDRTS with striatal involvement
The striatal variant was reported in 22 patients to date [9-13]. Patients showed early-onset and severe clinical manifestation including neonatal hypotonia, feeding difficulty and respiratory failure followed by short life expectancy. Ten of 22 patients had c.1771-6C>G variants, and 12 patients had c.1771-7C>G variants as either heterozygous or homozygous variants [9-13]. Our patient has a c.1771-6C>G variant and also showed profound neurodevelopmental problem and striatal involvement. It suggests genotype-phenotype association, as these variants were reported only in the striatal variant. Patients with theses variants shared MRI findings and relatively early-onset neurologic manifestations. The index patient, however, had clear differences with previous cases: lipodystrophy and progeroid facial appearance are key features of WDRTS and not reported in previous cases with striatal variant. Our case showed no dystonia or dyskinesia common neurologic symptoms in the striatal variant cases. Hiraide et al. [10] reported a patient with c.1771-6C>G, who showed mild developmental delay (walk independently at 1.5 years old) and striatal involvement. Therefore, further clinical observations and functional research are required to demonstrate detailed genotype-phenotype correlation.
In conclusion, we report an atypical case of POLR3A-related diseases. The patient was diagnosed with WDRTS by characteristic clinical features, and also had main MRI findings and neurologic features of the striatal variant with the c.1771-6C> G variant typical to the striatal variant of POLR3A-related disorders. As far as we know, this is the first WDRTS case in Korea. Also, this case is the first POLR3A-related disorder presenting the WDRTS and the striatal variant, which broadens clinical spectrum of the disease. Our case also strengthens the genotype association to the striatal variant, although further researches should be supported.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


