




Graphical abstract. ASD, autism spectrum disorder.
Introduction
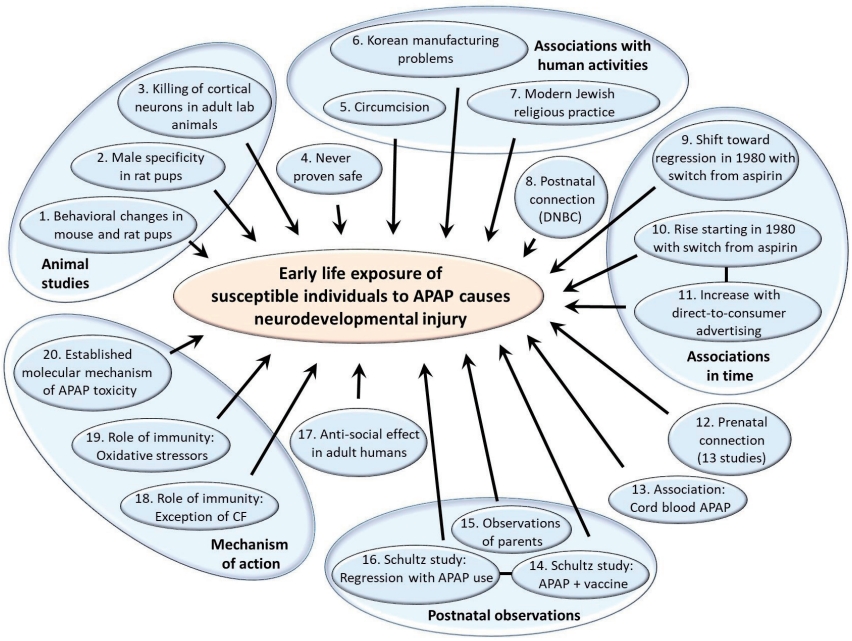
Acetaminophen (APAP; N-acetyl-p-aminophenol; paracetamol) as well as an antidote for its overdose (N-acetylcysteine) are listed by the World Health Organization as essential medicines for children [1]. Despite the worldwide acceptance of APAP in pediatric medicine, evidence that exposure to the drug during early development is a primary inducer of neurodevelopmental injury has been mounting for more than a decade. Although evidence is largely circumstantial or based on animal model studies, the preponderance of evidence weighs so heavily that a causal relationship can be inferred without remaining reasonable doubt [2]. This evidence is summarized in Fig. 1 and Table 1. Evidence demonstrates that, while most babies and children are relatively unharmed by APAP exposure, some are at risk due to the presence of oxidative stress [2,3]. Evidence points conclusively to the induction of autism spectrum disorder (ASD) with possible connections to both developmental delay and attention deficits [2]. Furthermore, evidence points toward exposure between birth and approximately 5 years of age as the period of highest risk, with the risk during prenatal exposure being significant in numerous studies [4] albeit less consequential [2,3]. Much of this evidence has recently been reviewed in detail elsewhere [2,3].
A recent exhaustive review of the literature complete with citation tracking demonstrated that, within the medical profession, APAP is widely considered safe when used as directed in the pediatric population [5]. Unfortunately, the widely held belief that it is safe for pediatric use is based on numerous clinical studies that assume that the liver is the target of drug toxicity [5]. In adults, the liver was identified as the target of APAP toxicity in the 1960s [6-8]. At that time, however, the view that babies metabolize drugs identically to adults was already known to be an unreliable and potentially dangerous assumption [9]; this knowledge had yet to be applied to the toxicity of APAP in children [5]. More than a decade later, a study using laboratory animals demonstrated that this assumption probably did not apply to APAP metabolism [10]. Although the target organ of APAP toxicity in newborn rats was not identified in that study, it was not the liver [10], a finding that was recently verified [11]. Within the last decade, the brain was identified as a target organ for APAP toxicity in newborn laboratory mice based on profound, long-term loss of cognitive function observed following exposure to relatively low drug doses [12]. Supporting the view that APAP is neurotoxic, a 2010 study of adult rats demonstrated that APAP induces the death of cortical neurons at concentrations lower than those required to induce acute liver failure [13].
A recent summary statement by Bauer et al. [14] examined the potential role of APAP exposure in utero in the induction of neurodevelopmental problems. This summary statement called for increased awareness of the potential role of APAP in this phenomenon, but it was criticized heavily by the American College of Obstetricians and Gynecologists (ACOG). In its response [15], the ACOG concluded that available studies “show no clear evidence that proves a direct relationship between the prudent use of APAP during any trimester and fetal developmental issues.” The ACOG further concluded that “physicians should not change clinical practice until definitive prospective research is done.” Considering the ACOG response, it is important to note that evidence of APAP inducing neurodevelopmental problems during the prenatal period is concerning but limited [2,16,17]. As shown in Table 1, only one of 20 lines of evidence was related only to the prenatal period. The other 19 lines of evidence were consistent with the involvement of the postnatal period and, in some cases, indicated that the postnatal period is the time of the greatest sensitivity to APAP induced neurodevelopmental injury. The relative safety of the prenatal versus postnatal period is perhaps not surprising given the particularly efficient metabolism of APAP by the mother during pregnancy [18] and the limited capacity of neonates to metabolize pharmaceuticals [19]. Unfortunately, considerable public debate concerning APAP-inuced neurodevelopmental problems has focused on the Bauer consensus statement involving the prenatal period [14] and is fueled by ongoing lawsuits involving its prenatal use [20]. Thus, despite substantial controversy surrounding the view that APAP causes neurodevelopmental injury in susceptible individuals, this debate and its surrounding controversy are primarily focused on the relatively limited evidence pointing specifically toward the prenatal period. Tragically, the public debate has not yet moved toward its postnatal use, for which relative and absolute risks are greater and evidence of neurodevelopmental injury is conclusive [2].
As shown inTable 1, evidence that early exposure toAPAP causes neurodevelopmental injury in susceptible babies and children is much more robust than that limited to prenatal exposure only (Table 1). The body of extensive evidence pointing to APAP-induced neurodevelopmental injury in the postnatal period has not been directly challenged. However, objections have been voiced to particular lines of evidence. Here, we review several issues that may be considered controversial in the field, considering each line of evidence independently and in the light of all other lines of evidence. In particular, issues associated with studies in humans and animal models and factors associated with APAP metabolism were considered.
Use of APAP in babies and small children was not monitored as medical practice changed
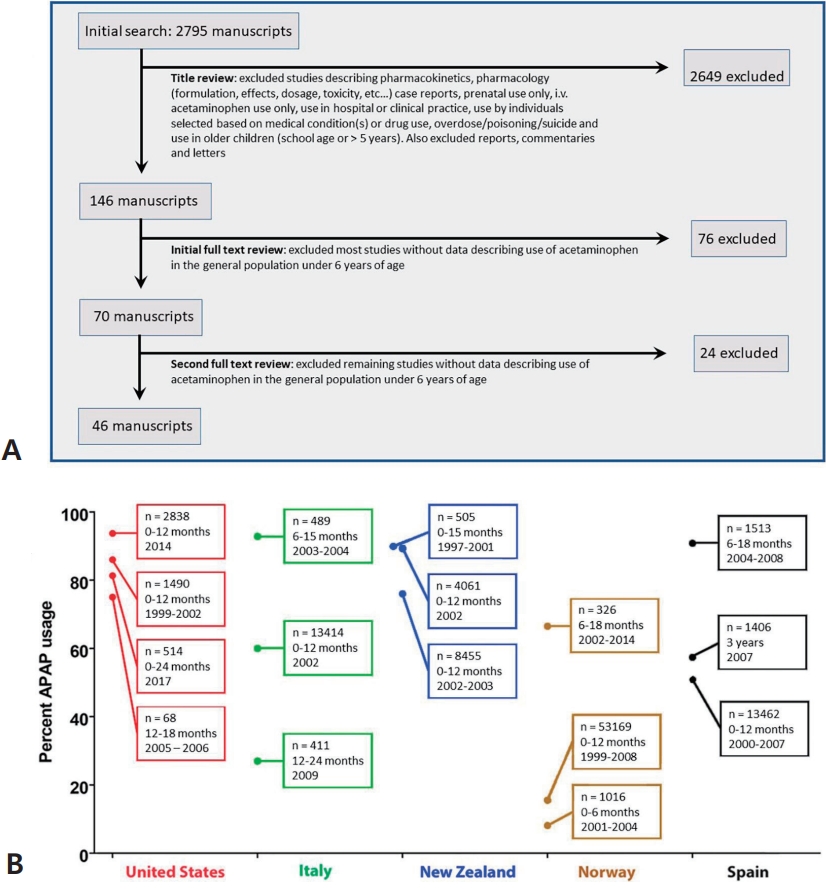
To establish any association between the use of APAP in the pediatric population and the incidence of neurodevelopmental disorders, it is most convenient to establish the prevalence of both factors over time with some degree of certainty. A systematic review was conducted to evaluate what is known about the prevalence of APAP use in the pediatric population at different times and locations (Fig. 2A). Although 48 studies were identified that evaluated the extent of APAP use in babies and children under 6 years of age, it is difficult to establish exactly how much APAP was used historically and when or where practice changed. Data were obtained from 38 countries, and those of 14 were limited to the International Study of Asthma and Allergies in Childhood (ISAAC) [21] in 2000–2003. Furthermore, in four countries in which the ISAAC study was not the lone study, its results deviated by a mean of 26.5% from those of other studies. In Hungary and Portugal, the ISAAC study found higher APAP use than other sources, whereas in New Zealand and Spain, the study found lower APAP use. In addition, results from the Danish National Birth Cohort (DNBC) [22] were also in disagreement with independently conducted work, with approximately 10% using APAP during the first 18 months of life [23] versus 65% using it within a 3-month period in an independent study evaluating a subset of the population assessed by the DNBC [24]. Furthermore, data from the Avon Longitudinal Study of Parents and Children study in England [25] were not consistently reported, with the use of APAP in babies 0–6 months of age during 1991–1992 reportedly 6% [26] and 84% [27].
Moreover, data from more than one independent study were found for only 11 of the 38 countries while three or more studies were found for only five countries. Fig. 2B shows the results from those five countries (the United States [US], Italy, New Zealand, Norway, and Spain), for which at least 3 independent studies evaluated the use of APAP in babies and children under 6 years of age. Although numerous studies have been conducted in various countries since the late 1990s, trends over time are not evident, and the results vary considerably. This makes it difficult to correlate changes in the prevalence of neurodevelopmental disorders with changes in medical practice. However, as discussed in the next section, key historical events affecting APAP use in the pediatric population have been documented that can be useful in estimating APAP use through time.
Associations between prevalence of ASD and early exposure to APAP
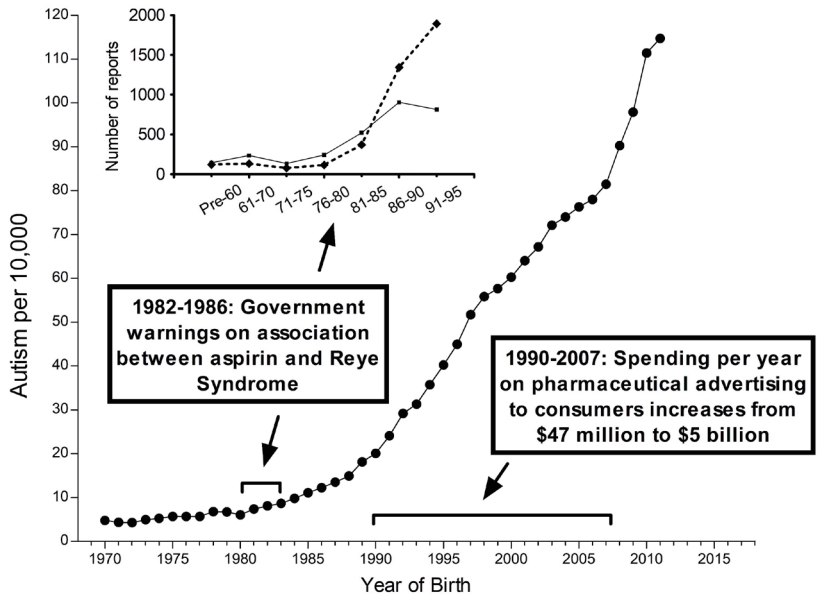
At least three of the 20 lines of evidence summarized in Table 1 and in Fig. 1 involve the association through time between factors affecting pediatric use of APAP and the prevalence of ASD (Fig. 3). One such temporal relationship (Fig. 3) entails an increase in the ratio of regressive versus infantile ASD beginning in children born after 1980 [28], which coincides with the time that aspirin use in babies and children was being replaced by APAP use due to increasing awareness of the connection between aspirin and Reye syndrome [29-31]. This shifting ratio indicates that some factor was introduced into the population that could induce ASD even after brain development had proceeded for years on a relatively normal trajectory.
A second distinct temporal relationship (Fig. 3) involves the beginning of the rise in the prevalence of ASD in the early 1980s, coinciding again with the replacement of aspirin in babies and children with APAP due to concerns over Reye syndrome. Although it has been argued that aspirin was replaced by ibuprofen rather than APAP in the US in the early 1980s [32], this is contradicted by available data demonstrating that APAP was the drug of choice in the US when the pediatric use of aspirin was dramatically reduced [29,30], Furthermore, as pointed out by Saugstad [33], ibuprofen was not approved as a prescription drug for children in the US until 1989, more than 30 years after a formulation of APAP was first marketed for children. Finally, ibuprofen was not approved for over-the-counter use in children until 1995 [34], long after the measured prevalence of ASD began to increase (Fig. 3).
A third temporal correlation is shown in Fig. 3 in which the rate of ASD continued to climb as direct-to-consumer advertising in the US increased dramatically and then became a part of US culture. However, the actual use of APAP in the pediatric population has been poorly tracked as discussed above. Trends in use over time are complicated by multiple means of acquiring the drug: through administration by physicians in clinics and hospitals and by caregivers using over-the-counter formulations at home. Thus, while changes in the quantity and qualitative nature of ASD coincide with major events affecting pediatric APAP use (Fig. 3), the exact pattern of change over time cannot be accurately ascertained from the literature. Nevertheless, the use of APAP in babies and young children, which was a relatively uncommon occurrence half a century ago, is now extremely common.
One potential argument that APAP cannot cause ASD is that the rising rates of ASD over time are, at least in part, a consequence of changing diagnostic criteria, increased awareness, and other factors (discussed by co-author CDN and colleagues [35]). Based on this argument, it was concluded that no chemical can account for the increased rate of ASD [36,37]. However, careful analysis of epidemiological evidence strongly suggests that the perceived increase in ASD since 1980 is real, at least in part, and not entirely due to artificial inflation [35]. Furthermore, the view that increases in the incidence of ASD are not real cannot readily account for the changing ratio of regressive to infantile ASD observed in the early 1980s (Fig. 3). Perhaps more importantly, disparities in the prevalence of ASD measured in side-by-side cohorts [38,39] demonstrate that some environmental factor or factors, at least under certain circumstances, play a pivotal role in the induction of ASD [2]. Finally, a number of factors independent of epidemiological evidence point toward a causal role of early exposure to APAP in the induction of neurodevelopmental disorders, particularly ASD [2,3].
Other objections to the conclusion that early exposure to APAP causes ASD in susceptible children include the fact that an association does not prove causation [40]. On the other hand, causation cannot exist without association, and multiple independent associations coupled with other lines of independent evidence support causation. However, temporal associations in this case were complicated by several factors. For example, as pointed out above, the actual use of APAP in the pediatric population was not tracked well over time. In addition, factors affecting oxidative stress, the necessary co-factor in APAP-induced neurological injury (discussed in detail below), may change over time [41]. Furthermore, the idea that medical establishments and society in general might need to recalibrate diagnostics and the awareness of a rapidly increasing incidence of cognitive dysfunction seems reasonable. Such a recalibration could account for short-term shifts in data concerning the incidence of ASD. Nevertheless, it seems implausible to attribute the dramatic and steady 40-year increase in prevalence to such factors. Indeed, ASD, although known by other labels over time [42], has consistently been distinguished by a deficit in social awareness [43] and was viewed as rare by knowledgeable individuals in the US and independently in Europe at the time of its discovery 80 years ago [44,45].
Studies in humans probing association between postnatal exposure to APAP and ASD
Limited studies have attempted to ascertain the association between postnatal exposure to APAP and ASD in humans. Notably, Alemany et al. [26] recently observed an increase in ASD associated with the postnatal use of APAP in the DNBC. The analysis showed an unacceptably large odds ratio (1.30) for a common occurrence (postnatal APAP exposure), indicating that the postnatal exposure to APAP reported in this study accounts for a substantial number of cases of ASD. However, despite the inclusion of more than 60,000 children, the degree of uncertainty ranged from an odds ratio of 1.02 (clinically insignificant) to 1.66 (intolerable by any standard). Thus, it is not possible to draw firm conclusions from the study of Alemany et al. [26] on the importance of postnatal exposure to APAP in the pathogenesis of ASD. We have previously demonstrated that the common use of the drug in babies and children without oxidative stress (and thus not at risk for APAP-associated neurodevelopmental problems) interferes with multivariate analyses, such as the one performed by Alemany et al. [26], resulting in (1) underestimation of the impact of APAP on the incidence ofASD and (2) a lack of statistical power leading to confidence intervals that are too large to draw conclusions [2]. Since the lack of reliability of the multivariate analysis in this context was examined previously [2], it will not be discussed here. An additional problem with the analysis of data obtained from databases, such as the DNBC, is evident in the systematic review described above. This review casts doubt on the reliability of information pertaining to APAP use in large databases, which could adversely affect the reliability of the results obtained from the data analysis. Thus, results from multivariate analyses of large datasets do not provide a valid basis for asserting that early exposure to APAP might be safe for neurodevelopment.
The first study to indicate that pediatric use of APAP is associated with ASD was a survey-based, case-controlled study published by Schultz et al. [46], a physician who saw his son develop regressive ASD following a vaccination [47]. Schultz et al. [46] noted that APAP use with vaccination was associated with ASD. In cases in which APAP was not administered, no significant association with ASD was found. The odds ratios for ASD diagnosis following APAP exposure were striking, depending on the comparisons made, exceeding 20-fold in some cases [46]. Although the study by Schultz et al. [46] was small, the results were persuasive and comprised one piece of evidence of early exposure to APAP as a cause of ASD [2].
Several criticisms of the study of Schultz et al. [46] have been published, some of which can be readily dismissed. For example, one objection was that Schultz et al. [46] did not “estimate a sample size required for a study of this nature (a survey study).” [48] In response, Schultz [49] stated that, given that calculating a study’s appropriate sample size requires some foreknowledge of the size of the expected effect, the appropriate sample size could not have been calculated prior to initiation of the study. The fact that the comparisons were statistically significant does, in fact, demonstrate that the sample size was adequate.
The most common objection to the study of Schultz et al. [46] is that the selection of subjects from internet groups produced a “biased sample.” [40,48] The supposition that the study of Schultz et al. [46] was undermined by bias among the participants may explain why the study never affected clinical practice, did not stimulate follow-up studies, and was omitted more than once during critical considerations of the role of APAP exposure in neurodevelopmental outcomes [32,50]. Given the potential importance of the study of Schultz et al. [46], it is worth examining the potential bias of the cohort studied. The cohort was recruited from 2 internet-based groups in 2005 and 2006 after both Wakefield et al. [51] and Rimland [28] suggested that vaccines might cause ASD. Furthermore, the bias that vaccines cause ASD has persisted in parents of children with ASD [52,53], so it seems highly likely that the parents in the study of Schultz et al. [46] were biased in favor of the view that vaccines can induce ASD.
In contrast to biases related to vaccines, a review of the literature published at the time suggests that bias probably did not exist favoring the view that early exposure to APAP causes ASD in susceptible children. A PubMed search using the terms “paracetamol” or “acetaminophen” and “autism” revealed only four papers prior to 2006. None of the four studies suggested that APAP might cause ASD. The initial study, by Alberti et al. [54] in Italy, showed profound impairment of APAP metabolism in children with ASD and was published in 1999, several years prior to the study of Schultz et al. [46] However, Alberti et al. [54] did not suggest that exposure to APAP causes ASD. Furthermore, the study of Alberti et al. [54] was cited in PubMed-indexed journals only 3 times prior to 2006 [55-57], all within the context of understanding the physiology of ASD, not the cause. The study of Alberti et al. [54] was cited in the Alternative Medicine Review (not PubMed indexed) in 2002 [58], and APAP was listed by the author as a potentially neurotoxic compound in children with oxidative stress. However, concerns regarding APAP occupied only one line of a 25-page report that included a page-long discussion on the potential role of vaccines and vaccine components in the induction of ASD. In 2003, Torres at the Utah State University suggested that the use of antipyretics in general may lead to ASD [59]; however, the hypothesis was that the absence of fever, rather than the presence of APAP, might be a problem. This paper was not cited in the literature until 2009, and was, interestingly, cited in the context of the potential importance of vaccines, not APAP, in the etiology of ASD [60]. Furthermore, coauthor WP has been actively engaged with the community of parents of children with ASD and has observed that few parents, even in the past 5 years, have been aware of the view that early exposure to APAP can cause ASD in susceptible babies and children.
Thus, it seems highly likely that the parents surveyed in the study of Schultz et al. [46] were indeed biased in favor of the idea that vaccines cause ASD; however, it seems unlikely that they had a similar bias against APAP. Indeed, as Schultz explained, “The hypothesis that APAP causes ASD was completely unknown to the parents being surveyed. In fact, my study conducted in 2005 and 2006 was the first to explore this hypothesis.” (personal communication with coauthor WP, used with written permission.) It has been suggested that parents with ASD might try harder to recall information while searching for answers [40]; however, studies probing this issue have not found that parents of children with adverse outcomes have better recall [61]. Perhaps more importantly, the data provided by Schultz et al. [46] do not suggest that parents of children with ASD have better recall than parents of neurotypical children. The study of Schultz et al. [46] used yes or no questions, and the response rate to particular questions could be taken, at least in part, as a surrogate indicator of recall. Using the response rate as a metric, the data of Schultz et al. [46] revealed no evidence that parents of children with ASD had better recall than parents of neurotypical controls. For example, the answer rate was 100.0% for cases and controls when asked about their child’s APAP use in conjunction with vaccines, 83.1% and 85% for cases and controls, respectively, when asked about their child’s APAP use between 12 and 18 months of age, and 59.0% and 72.5% for cases and controls, respectively, when asked about their child’s exposure to ibuprofen between 12 and 18 months of age. Furthermore, Schultz specifically addressed the issue of recall by independently analyzing surveys with a greater time lapse since the events in question. As pointed out by Schultz [49], the results were robust and did not indicate that time had affected the outcome.
With the above discussion in mind, the conclusions of the study of Schultz et al. [46] can be amended. In cases in which the parents are likely biased toward the view that vaccines cause ASD, exposure to APAP rather than vaccines was likely a factor in the induction of ASD in their child. Furthermore, it is apparent that the dismissal of the study due to bias is unwarranted and not supported by any available information. Thus, the study of Schultz et al. [46] contributed evidence pointing to APAP use as a cause of neurodevelopmental injury in susceptible babies and children [2].
Clues from APAP metabolism
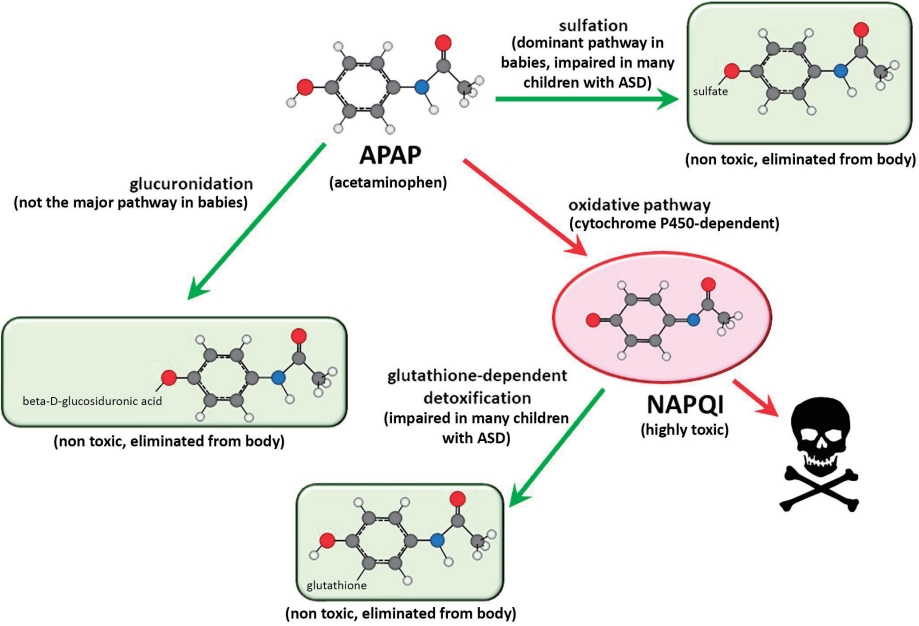
The metabolism of APAP is well characterized and provides considerable insight into how APAP can cause neurodevelopmental injury [41,62]. The human body processes APAP via three primary pathways (Fig. 4). Two of these pathways involve the addition of highly water-soluble structures: glucuronate via the glucuronide pathway or sulfate via the sulfation pathway. In adults, the addition of glucuronate predominates over the addition of sulfate [63], whereas in babies and children under the age of 9 years, the addition of sulfate predominates over the addition of glucuronate [63,64]. The third pathway also involves the addition of a highly water-soluble molecule (glutathione). The first step of this pathway involves the production of a highly toxic substance, N-acetyl-p-benzoquinone imine (NAPQI). Fortunately, in healthy individuals, NAPQI is rapidly neutralized by glutathione (Fig. 4). Unfortunately, children with ASD tend to have an impaired ability to utilize the sulfate pathway [54,65,66]. Additionally, children with ASD tend to experience oxidative stress [3,67], which depletes glutathione [65]. Furthermore, APAP exposure significantly depletes glutathione [68], suggesting that repeated exposure to the drug is potentially more hazardous than a single exposure.
Although APAP metabolism is well characterized, a high degree of variability in APAP metabolism within the pediatric population has been observed, and the factors affecting this variability are poorly understood [69]. One factor affecting the metabolism of APAP is the presence of autoantibodies that impair folate transport to the brain, which is found in almost 3 quarters of children with ASD [70]. Since folate is necessary for the synthesis of glutathione, an impaired ability to detoxify NAPQI is expected in these children. Thus, many children with ASD have impaired sulfation and glutathione-dependent pathways for the clearance of APAP. A third pathway, glucuronidation, has been speculated to compensate for this problem [40]. However, the glucuronidation pathway is not upregulated by repeated exposure to APAP [71] and is a minor pathway in babies and children, as discussed above [63,64]. Furthermore, because some NAPQI is created regardless of the function of the other 2 pathways, failure in the glutathione-dependent pathway is expected to result in the accumulation of NAPQI and subsequent toxicity, even if the other 2 pathways are functional.
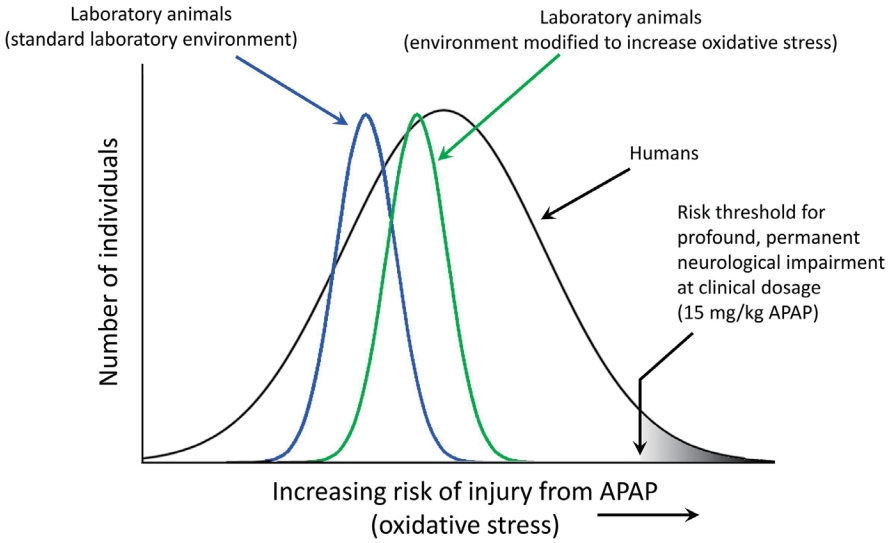
It is not surprising that both sulfation- and glutathione-dependent pathways are aberrant in the same population because they are metabolically connected [41,72,73]. Alterations in both pathways enhance oxidative stress and increase APAP toxicity. Unfortunately, even at levels of APAP that are currently considered acceptable, this situation will result in the exposure of some babies and children to levels of APAP toxicity that are much greater than those seen in typical, nonsusceptible individuals or in laboratory animals exposed to the same doses of APAP (Fig. 5).
Discussion
In this narrative review and our previous narrative reviews on the safety of pediatric APAP use, we addressed several lines of evidence that might be considered controversial. We believe that considering multiple lives of evidence is necessary given the complexities particular to this topic. For example, a recent systematic review and meta-analysis by Tan et al. [74] at the University of Auckland considering almost 20 studies and a quarter of a million children less than 2 years of age raised no substantial flags concerning the safety of early exposure to APAP. Unfortunately, based on the approach used in the study by Tan et al. [74], the results obtained were expected regardless of whether early exposure to APAP was responsible for most cases of ASD. Tan et al. [74] noted that exposure rates to APAP in the pediatric population now approach 95%, a factor that precludes the identification of APAP as a causative agent in neurodevelopmental disorders using a multivariate analysis of large data sets [2]. Consistent with our recent results [5], Tan et al. [74] noted that the measures of adverse outcomes were limited to acute events rather than neurodevelopmental outcomes. As previously discussed, other factors impede the usefulness of such analyses, including the need for long-term monitoring of exposure from the time of conception, the inability to separate confounding factors from oxidative stress-inducing cofactors, and the use of intravenous formulations of APAP containing an antidote for toxicity in some studies. Indeed, an evaluation of the effect of early exposure to APAP on neurodevelopmental outcomes would require substantial effort that is unlikely to occur in the near future as previously discussed [5].
Studies in animal models are currently sufficient to conclude that early exposure to APAP causes neurodevelopmental problems [5]. The observation of APAP-induced neurodevelopmental problems in laboratory animals is robust, encompassing both laboratory rats and mice and a variety of study designs (see Patel et al. [2] and recent studies from the University of New Orleans [75,76]). However, studies have yet to recapitulate the symptoms of ASD, which remains a highly laudable goal of research in the field. Although it has been argued that “clinically relevant” doses of APAP should be used in such studies, it is expected that recapitulating conditions in susceptible humans using healthy laboratory animals will require higher drug doses than those commonly encountered by humans (Fig. 5). In summary, laboratory rats under ideal laboratory conditions will be more resistant to APAP-induced neurodevelopmental injury than humans that have significant problems metabolizing the drug. Not only are laboratory rats bred to be healthy under standard laboratory conditions, potentially reducing the genetic factors making them susceptible to disease, they are also fed an exceedingly healthy diet [2] and are often largely free of infections, environmental toxins, and other oxidative stress factors associated with ASD in humans. Taking this into account, current regulations stipulate that preclinical testing should include higher drug doses than those expected to be encountered by patients [77].
The failure of the medical community to accurately track APAP use in the pediatric population over time as well as its almost ubiquitous use identified in some studies (Fig. 2) reflects a high degree of acceptance of the drug. The incorrect assumption that babies react to APAP similarly to adults is a key factor in its current level of acceptance [5]. However, other factors undoubtedly contribute to this. For example, (1) critical studies in laboratory animals were conducted only recently; (2) most babies and children suffer no apparent serious adverse neurodevelopmental effects from APAP use; (3) severe adverse neurodevelopmental effects may not be diagnosed until long after drug exposure; (4) the diverse array of oxidative stress-inducing cofactors in APAP-induced neurodevelopmental injury creates a large and potentially confusing number of associations with neurodevelopmental injury; and (5) any severe adverse neurodevelopmental effects might be attributed to the indication for the drug.
Most clinicians and caregivers are not currently aware of the available knowledge concerning apparent adverse reactions to early APAP exposure in susceptible children. Conducting large long-term studies in human children may not be feasible as discussed above. However, this point may be irrelevant given that the preponderance of available evidence renders such a study unnecessarily risky and thus unethical. With this in mind, regulatory agencies and professional medical societies should move forward with the currently available information. The immediate goals are to first acknowledge and then promote awareness of the problem. Changes in medical practice should be implemented that effectively weigh the risks and benefits of neonatal and pediatric APAP use. Failure to implement change in medical practice currently constitutes disregard for the ample evidence of harm despite the absence of any valid rationale for the view that APAP might be safe for neurodevelopment. Finally, the ability of antidotes for APAP toxicity, such as N-acetylcysteine, to prevent APAP-induced neurodevelopmental injury could be probed.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


