




Introduction
C3 glomerulopathy (C3G) characterized by deposition of isolated or dominant C3 in the glomeruli, is a heterogeneous disease with complex triggering events and abnormalities of the alternative pathway of complement system [1,2]. The clinical diversity, histological profile, and kidney outcome of C3G in children is varied with clinical presentation and severity ranging from subclinical proteinuria or microhematuria to rapidly progressive glomerulonephritis (RPGN) and, occasionally chronic kidney disease (CKD) progressing to end-stage kidney disease (ESKD) [3]. There is a lack of consensus on the optimal diagnostic evaluation and treatment approaches in children with C3G.
Apart from histology and immunofluorescence study of kidney biopsy, other evaluations in C3G such as electron microscopy, serological tests of the complement pathway and genetic testing help in targeted therapies and prognostication [4,5]. However, these evaluations in resource-limited settings remain a challenge and are often incomplete due to either unavailability of advanced complement tests such as assays for C3 or C5 nephritic factor or inaccessibility to electron microscopy and genetic testing due to financial constraints. Newer therapies, such as eculizumab (C5a inhibitor), targeting the alternative, lectin and terminal complement pathways that have shown some benefit are difficult to procure [2,6-8]. Whether this impacts outcome is unclear.
The objective of this study was to describe the clinicopathological features of C3G, response to empirical immunosuppression and to evaluate the short-term kidney outcomes in children in a resource-limited setting.
Methods
In this retrospective cohort study, review of hospital case records of children diagnosed as C3G between January 2013 to December 2021 was performed after obtaining ethical approval of St. John's Medical College Hospital (IEC Number–159/2019). Children aged less than 18 years with kidney biopsy-proven diagnosis of C3G (dominant C3 staining, with intensity of ≥2 orders of magnitude more than any other immunoglobulin) were included, while those with post streptococcal glomerulonephritis (typical clinical features and/or positive antistreptolysin O antibodies titres) and immune complex-membranoproliferative glomerulonephritis (MPGN) pattern secondary to viral infections like Hepatitis B, Hepatitis C or Lupus nephritis were excluded. Demographic details, clinical features and laboratory findings at initial presentation, immunosuppressive therapy and kidney status during follow-up were noted.
The total number of pediatric kidney biopsies performed in the center during this period was 825, of which 46 (5.6%) were diagnosed as C3G. Based on electron microscopy, C3G was further subclassified as dense deposit disease (DDD) if features of highly electron-dense, osmiophilic ribbon-like intramembranous deposits were noted and C3 glomerulonephritis (C3GN) in the presence of discrete mesangial and capillary wall deposits of lesser intensity [9].
The clinical presentation at time of diagnosis was classified as nephrotic syndrome, acute nephritic syndrome, and RPGN using standard criteria. Nephrotic syndrome was defined as presence of nephrotic-range proteinuria (urine protein-creatinine ratio (uPCR) ≥2 or 24-hour urine protein excretion ≥40 mg/m2/hr) with hypoalbuminemia (serum albumin <3 g/dL) with or without edema. Acute nephritic syndrome was defined as the presence of gross or microscopic hematuria (≥5 red blood cells/high power field of centrifuged urine), hypertension (systolic or diastolic blood pressure ≥95th centile for age, sex and height), proteinuria and kidney dysfunction. Acute kidney injury (AKI) was defined as per KDIGO (Kidney Disease: Improving Global Outcomes) guidelines [10].
RPGN was defined as children with nephritis and rapid decline in kidney function, with doubling of serum creatinine over days to weeks with or without presence of crescents on histopathology [9].
Light microscopy, immunofluorescence patterns and electron microscopy findings (when available) of kidney biopsy specimens were analyzed. The histopathological pattern of injury on light microscopy was classified based on the characteristic findings as follows: MPGN – endocapillary proliferation, often diffuse and global, with double contours of glomerular basement membrane on silver stain; mesangioproliferative glomerulonephritis (MesPGN) – mesangial hypercellularity and expanded mesangial areas; diffuse proliferative glomerulonephritis (DPGN) – endocapillary or mesangial proliferation with inflammatory cell infiltrate and no crescents; focal crescentic glomerulonephritis – presence of crescents (extracapillary proliferation of more than 2 cell layers occupying at least 10% of the circumference of the glomerulus) in 10%– 50% glomeruli; crescentic glomerulonephritis – more than 50% glomeruli with crescents and thrombotic microangiopathy – fibrin thrombi, mesangiolysis, endothelial swelling, intimal proliferation of arterioles with thrombi [9,11-13]. Evidence of sclerosis (absent, <50% or >50%) and tubulointerstitial features of interstitial fibrosis and tubular atrophy (graded as absent, 1%–25%, 26%–50% and >50%) were noted. Intensity (≥2 orders of magnitude) and location (mesangial or capillary wall) of staining of C3 was recorded. Presence of other immunoglobulins (IgG, IgA, IgM, and C1q) were graded from 0 to 3+. Electron microscopy details included degree of podocyte effacement, presence of intramembranous deposits, location and, type of immune complex deposits.
The treatment protocol for C3G was individualized based on clinical presentation. All children received supportive treatment in the form of antihypertensives and/or antiproteinuric medications. Immunosuppressive therapy was initiated based on clinical features, laboratory parameters and histopathological findings. Pulse intravenous steroids (10–15 mg/kg/day for 3 days) and cyclophosphamide (500–750 mg/m2/dose, 6 monthly doses) were administered to those with RPGN or any percentage of crescents in biopsy irrespective of the severity of clinical presentation. Children with nephrotic syndrome or nephrotic-range proteinuria received oral steroids (1–2 mg/kg/day) and additional mycophenolate (MPA) (600–1,200 mg/m2/day), if response to steroids was inadequate [3,9,14]. All children were advised follow-up once every 1–3 months, based on their clinical status.
The kidney outcome measures were classified as having (1) ‘complete kidney recovery’ if normal kidney functions (estimated glomerular filtration rate [eGFR] ≥90 mL/min/1.73 m2), reduction in proteinuria to <0.3 g/day or spot uPCR <0.2, absence of microscopic hematuria and no documented hypertension during clinic visits; and (2) ‘kidney sequelae’ if there was a need for dialysis, serum creatinine more than 30% of baseline, proteinuria ≥0.3 g/day or spot uPCR ≥0.2, hematuria or hypertension. Relapse was defined as recurrence of proteinuria (≥0.3 g/day or spot uPCR ≥0.2), hematuria or abnormal kidney function (serum creatinine more than 30% of baseline) after attaining complete kidney recovery.
The statistical analysis was carried out using IBM SPSS Statistics ver. 19.0 (IBM Co., Armonk, NY, USA). Categorical variables were expressed as frequency and percentages. Continuous variables were expressed as mean±standard deviation (SD), or median and interquartile range (IQR) as appropriate.
Results
The mean±SD age at diagnosis in the cohort (n=46) was 9±4 years, majority (33, 71.7%) being boys. The clinical features are presented in Table 1. The most frequent clinical presentation was acute nephritis (27, 58.6%) while nephrotic syndrome was observed in 10 (21.7%). Hypertension and hematuria were seen in 42 (91.3%) and 40 (87.0%) respectively. A history of upper respiratory tract symptoms and mild fever, was present in 36 of 46 children (78.2%). The mean±SD C3 levels were 58.4±48.9 mg/dL with low C3 levels noted in most (35, 76.1%) children, while C4 levels were also low in 9 (25%. n=36). C3 nephritic assay was not done as assay is unavailable in the country. None of children underwent genetic testing for variants in gene regulating the alternate complement pathway.
The median (IQR) eGFR was 46.8(16.1, 78.9) mL/min/1.73 m2, 28 (60.1%) children had eGFR lesser than 60 mL/min/1.73 m2. AKI was present in 22 (47.8%) and RPGN was noted in 9 (19.5%). Kidney replacement therapy (KRT) was required in 10 children (21.7%) at time of diagnosis.
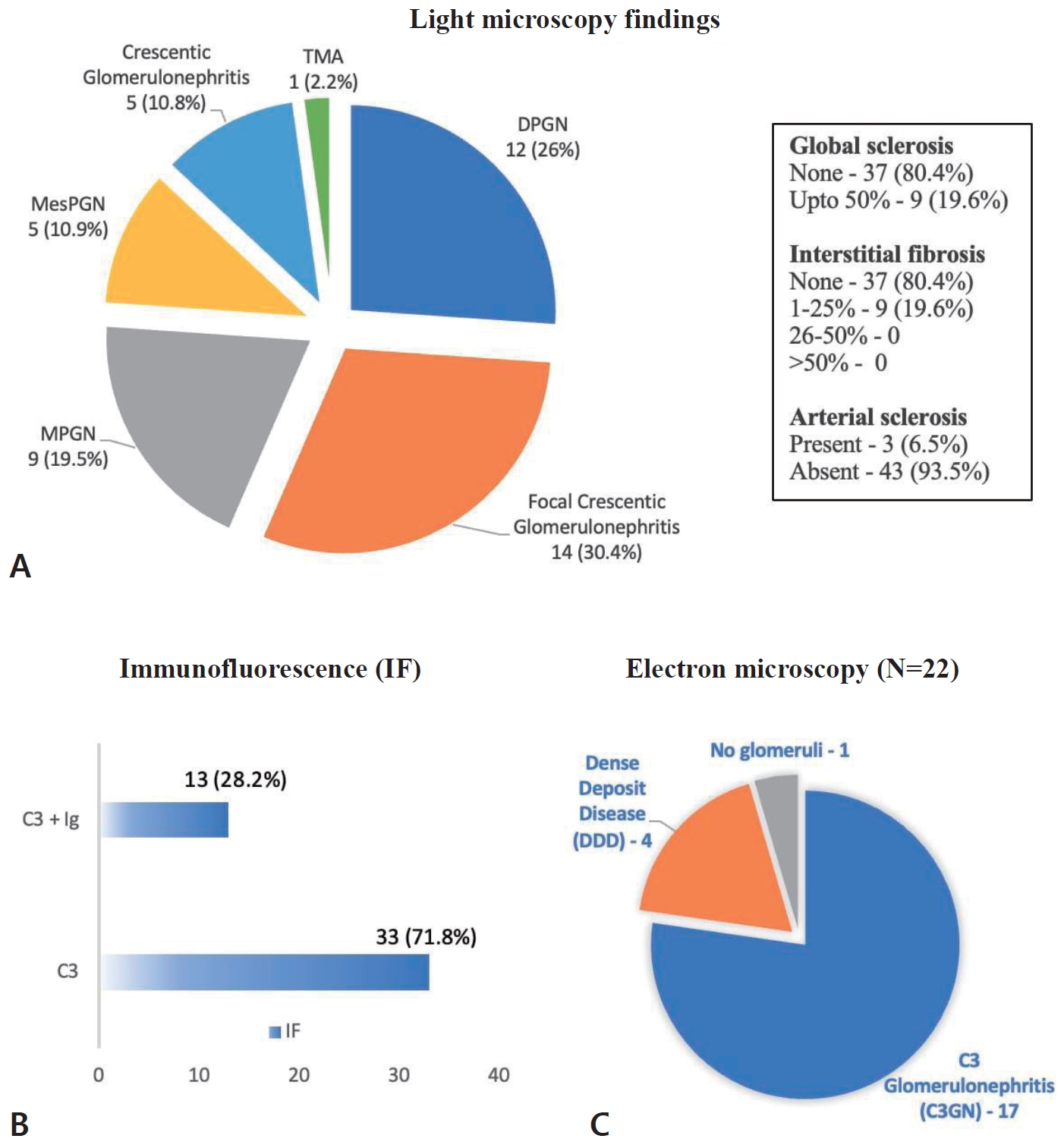
Focal crescentic glomerulonephritis (14, 30.4%) and DPGN (12, 26%) were the most common histological patterns of injury noted in kidney biopsy (Supplementary Fig. 1). Global sclerosis up to 50% was observed in 9 (19.6%), while none had interstitial fibrosis >25%. Arterial sclerosis was observed in only 3 (6.5%) of the biopsies.
Immunofluorescence revealed isolated C3 deposits in 33 (71.8%), while one-fourth (13, 28.2%) showed dominant C3 with other immunoglobulin staining.
Electron microscopy was performed in less than half (22, 47.8%) of the cohort who could afford, of whom 17 (77.3%) were classified as C3GN and 4 (18.2%) as DDD (there was no glomerulus in the tissue sent for electron microscopy [EM] in 1 patient). Fig. 1 highlights the histopathological characteristics of the cohort.
Almost two-thirds (n=29, 63%) received empirical immunosuppression therapy at onset. All received oral prednisolone, while intravenous methylprednisolone therapy was used in 13 children (28.2%). In addition to steroid therapy, 9 children (19.6%) also received intravenous cyclophosphamide while MPA was started in 2 children (4.3%).
Among the 42 (91.3%) who were noted to be hypertensive at onset, 37 (88%) were discharged on oral antihypertensives. Antiproteinuric therapy with angiotensin converting enzyme inhibitors were prescribed in 7 (15.2%) once serum creatinine normalized.
Hemodialysis was required in 10 children (21.7%) with half of those with DDD requiring dialysis at time of diagnosis. At discharge, one-third (15, 33.6%) continued to have abnormal creatinine and 5 (10.8%) were dialysis dependent (Table 1).
The median (IQR) duration of follow-up was 11.5 (6–24 months) months with 31 (67.4 %) having at least 1 follow-up after discharge from hospital. C3 levels normalized in 30%, while hypertension, proteinuria and microscopic hematuria persisted in 71%, 51% and 60% of children respectively. During follow-up, MPA was used in 8 children (25.8%) (in 2 of them as maintenance immunosuppression post cyclophosphamide), 1 (3.3%) received calcineurin inhibitor (due to MPA failure) and 1 (3.3%) received rituximab (due to poor response with cyclophosphamide). The immunosuppressive therapy and response are highlighted in Table 2.
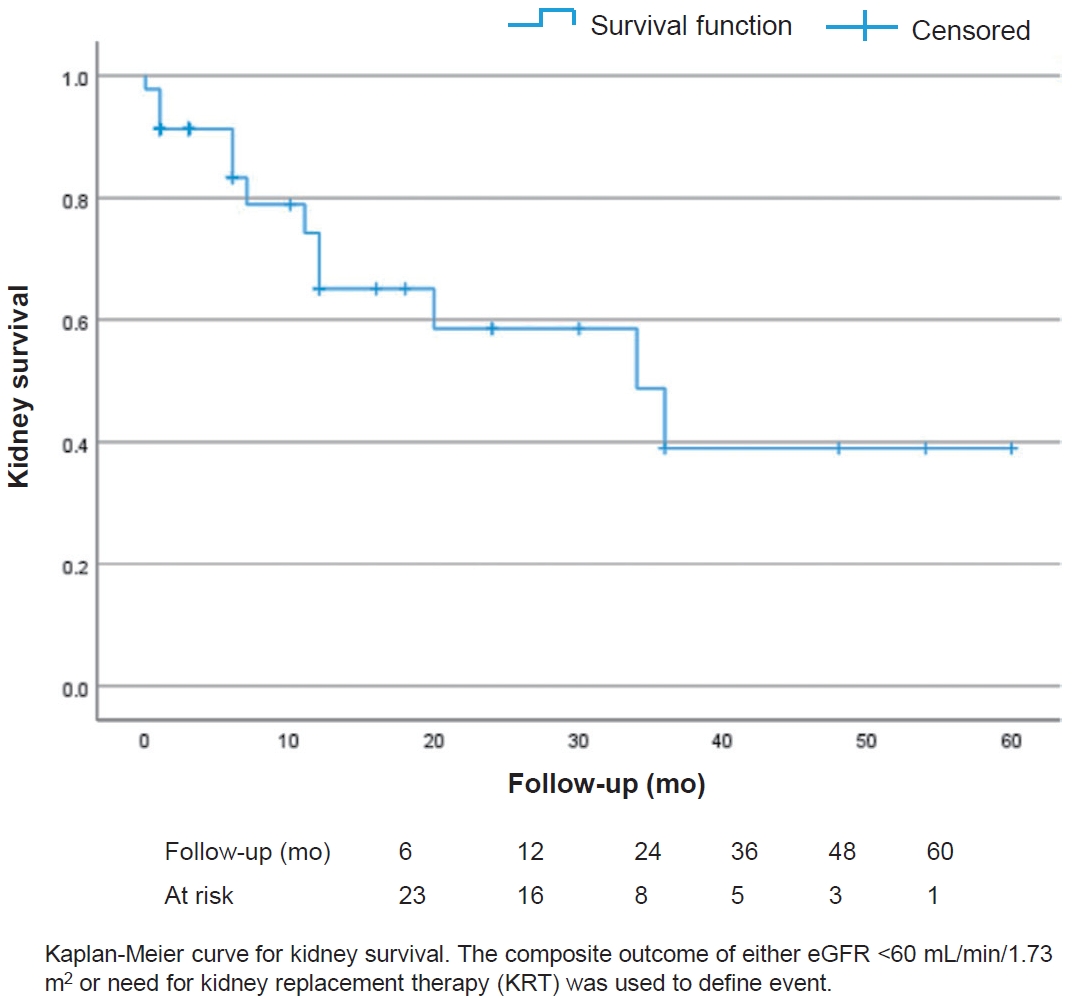
The kidney survival (i.e., event free follow-up without eGFR <60 mL/min/1.73 m2 or need for KRT) decreased over time in the total cohort (Fig. 2). Small sample size precluded a comparative analysis of kidney survival in patients with C3GN compared with DDD. At the last visit, among those with at least 3 months of follow-up, complete kidney recovery was seen in 6 children (19.3%), while 25 (80.6%) continued to have kidney sequelae of whom 7 (23.3%) progressed to CKD despite receiving immunosuppressive therapy. Among those who had complete kidney recovery none had relapse till the last follow-up. Of the 5 children (16.1%) who progressed to ESKD, 4 required maintenance dialysis therapy. Two children (4.3%) died during the follow-up period. The cause of death in these 2 children were acute pulmonary edema secondary to hypertensive cardiac failure and catheter related blood stream infection.
Discussion
This study describes the clinical spectrum and outcomes of children diagnosed with C3G in a resource-limited setting. C3G was commonly seen in older children and associated with a variable clinicopathological spectrum. Majority presented with glomerulonephritis with significant morbidity and one in 5 required dialysis at diagnosis. Immunosuppressive therapy resulted in complete remission in only small proportion. Although confounded by significant loss to follow-up, similar to previous reports from India [15,16], over two-thirds had persistent kidney sequelae, quarter of whom progressed to CKD. Comparison of findings with other studies of children with C3G is represented in Table 3.
The clinical presentation of C3G is heterogenous, with nephrotic syndrome reported as the commonest presentation (38%–77%) in most adult and pediatric C3G series [7,15,17-20]. In this study, the predominant presentation was acute nephritis, similar to another study, although 28.2% among them also had nephrotic-range proteinuria [21]. Consistent to other pediatric studies, one-fifth presented with RPGN in the present study [20]. Of the small proportion of children (with available data) who had DDD, half of them had RPGN. As described in previous studies, this study also found an infection trigger at onset in the majority [19,20]. As reported in adult and pediatric studies, hypocomplementemia is not seen in all children with C3G (range of 40%–80%), and persisting low C3 observed in current study has also been observed in other studies as well (40%–60%) [8,15,20,22].
The probable mechanisms for low serum C4 levels noted in this study are presumed to be due to classical complement pathway convertase dysregulation or an infectious trigger and has also been reported in other similar studies [2,15,23-25]. Lack of availability of complement testing precluded further analysis.
Based on the recent classification [11], we found focal crescentic glomerulonephritis as the commonest histopathological pattern, which is in contrast to other pediatric series which have reported MPGN (37%–75%) and MesPGN (30%–60%) as most common biopsy features [10,15,19-22]. RPGN is associated with poor prognosis and is important to identify. One of the challenges faced in resource-limited settings is the lack of access to electron microscopy, due to high costs, which hinders the subclassification into C3GN and DDD. In our study we noted DDD in 4 of 22 (18.1%) and extrapolated the prevalence to be around 17%–20%, which is lower than the reported incidence of DDD (64.8 %) in a larger cohort of C3G from the same country [20]. The higher proportion of DDD in that study could be attributed to the center being a referral unit with easy access to electron microscopy. Identifying the subgroup of DDD is important as previous adult and pediatric studies have reported poor outcomes and need for aggressive management and close monitoring [7,15,20,21]. However, a recent study found no significant difference between children with DDD and C3GN in the clinical presentation or histological pattern [8]. In the present study, among the children diagnosed as DDD, half presented with RPGN, majority had crescentic glomerulonephritis pattern and experienced significant morbidity (50% required KRT) and mortality (50%). The small numbers precluded further analysis between C3GN and DDD in our study.
Genetic and complement pathway testing helps distinguish autoimmune and genetic forms of C3G which can help optimize management including prognostication and also help in donor selection for kidney transplant. The yield of genetic testing in pediatric studies is 9%–10%. Serological complement pathway tests may be useful markers to predict and monitor response to treatment as the prevalence of autoantibodies was found to be 46% (C3 nephritic factor – 39%, anti-CFH – 17%, antibodies to C3b – 7%) in a study that included 39 children with C3G [17]. Though some studies have shown lack of response to immunosuppressive therapy in those with autoantibodies and pathogenic genetic variants, whether lack of performing complement factor assays and genetic testing for complement regulatory genes in view of nonavailability and non-affordability of such advanced tests in the study impacted the outcome remains uncertain [7,17,22].
The current management reflects heterogeneity and lack of consensus on the treatment of C3G patients. Given the central role of complement pathway dysregulation in the pathogenesis of C3G, the most rationale treatment would be targeted complement therapy. However, there is insufficient evidence on the efficacy of complement inhibition with eculizumab, with reports in adults and children not showing any benefit [4,6,26].
Steroids are the mainstay of treatment and additional immunosuppressants like MPA, cyclophosphamide and tacrolimus have been tried with unpredictable response (remission in 11%–50%). The remission rate observed in other studies is in contrast to the current study wherein among those who received immunosuppression, only 12.5% attained remission while the majority had kidney sequelae. One-third of our cohort did not receive immunosuppression at initial presentation either due to mild clinical presentation (normal kidney functions/non-nephrotic proteinuria) or having persistent kidney dysfunction (eGFR <30 mL/min/1.73 m2). However, it is interesting to note that among these children a few (17.6%) attained remission with supportive therapy indicating that immunosuppressive therapy may not be indicated in all. In a recent pediatric series, MPA was the initial therapy in 62% among whom 27.5% attained complete or partial remission, while 50% of those treated with tacrolimus (13.5%) showed clinical response [20]. Another study in children showed remission in 58.9% with only steroid therapy [15]. In the present study, significant morbidity was seen at presentation with one-fourth requiring dialysis and a majority continued to have kidney sequalae with progression to ESKD in almost one-fifth. It was challenging to compare outcomes as the significant heterogeneity of treatment and lack of C3 nephritic factor assay/genetic testing in our study confounds outcomes. Nevertheless, the clinical response to therapy was worse compared to studies from high-income countries [7,8,17].
Some of the limitations of the study include unavailability of EM in more than half the cohort, and limitations of retrospective analysis. The significant loss to follow-up, reflecting the real-world scenario, probably due to low socioeconomic status, lack of universal healthcare coverage and poor health literacy, may be confounding the outcomes reported in this study.
In conclusion, pediatric C3G has a wide spectrum of clinical presentation and histopathological features, significant morbidity and persistent kidney sequelae. The diagnostic work-up and management of C3G in children in resource-limited settings is challenging due to lack of access to and nonavailability of sophisticated tests and targeted therapy. These factors pose a barrier to comprehensive phenotyping and management of C3G.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


