

Introduction
In 1963 Rubinstein and Taybi first delineated a syndrome characterized by broad thumbs, broad toes, mental and motor retardation, cardiovascular and renal anomalies, a beaked nose, and a downward slanting palpebral fissure1). Rubinstein-Taybi syndrome (RTS) includes a variety of cardiac problems such as an atrial septal defect, a ventricular septal defect, a patent ductus arteriosus (PDA), coarctation of the aorta, pulmonary stenosis and so forth. Pulmonary hypertension from cardiac anomalies had been reported in children with RTS2,3). Children with RTS can have upper airway obstruction during sleep due to hypotonia and the anatomy of the oropharynx and airway, which make these patients susceptible to obstructive sleep apnea (OSA)4,5).
We report here on a case of a patient with RTS and pulmonary hypertension, and the latter was due to congenital heart defects and OSA successively during infancy.
Case report
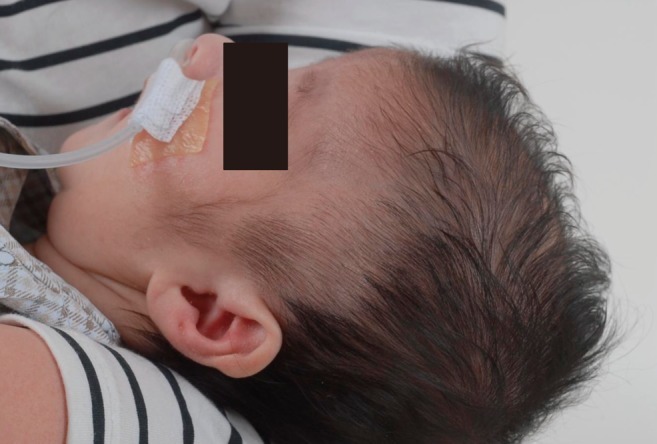
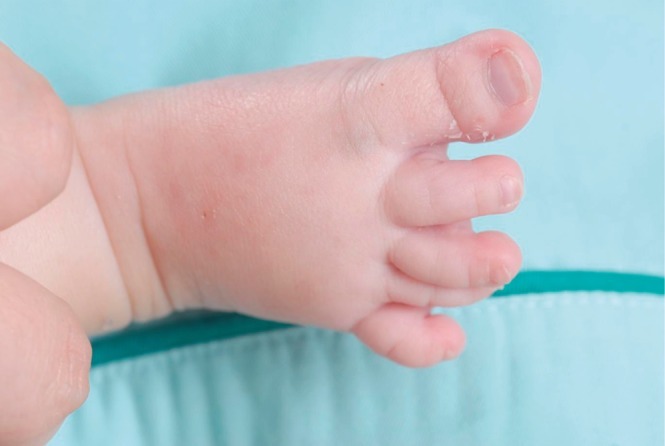
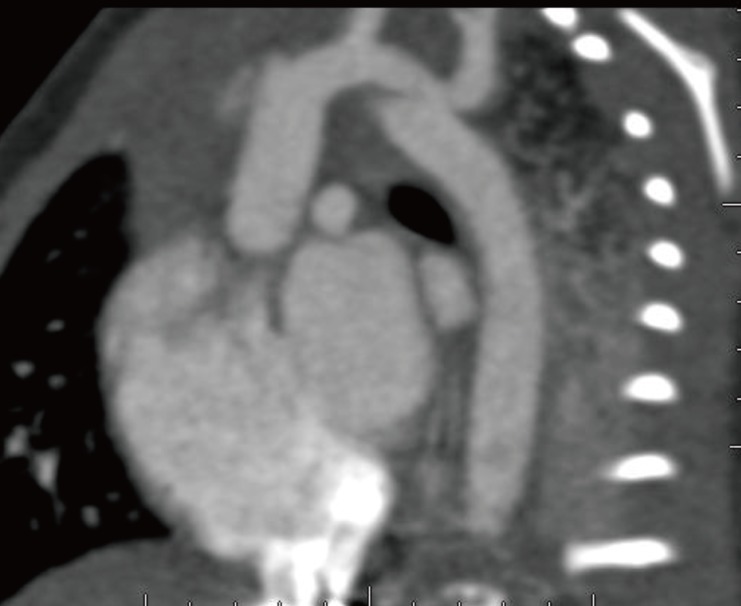
This male infant was born at 38 weeks 6 days gestation by Cesarean section. The Apgar score was 6 at one minute and 7 at five minute, and the birth weight was 3,300 g. The physical findings pertinent to RTS were broad thumbs with radial deviation, broad big toes (Fig. 1), a beaked nose with broad columella (Fig. 2) and a grade 3/6 systolic murmur. The echocardiogram showed a large PDA and a relatively small aortic arch. The PDA flow was bidirectional. Heart computed tomography showed arch hypoplasia, a large PDA and right ventricular dilatation (Fig. 3). The peak velocities of tricuspid regurgitation on the echocardiography that was performed weekly ranged from 3.6 to 4.6 m/sec before a corrective operation for the arch hypoplasia and PDA at 23 days after birth. The peak velocity of tricuspid regurgitation was decreased to 2.2 m/sec immediately after the operation, but this increased to 3.5 m/sec at 2 months later. Poor oral feeding, cold sweating and vomiting were noted at that time. After re-admission (1 month later after discharge from the hospital), we administered bosentan and intermittent oxygen under the impression of delayed pulmonary vascular remodeling. The general condition of patient was not improved and the oxygen saturation frequently fell below 90%, especially during sleep. Loud snoring, excessive sweating and recurrent respiratory pause were detected during sleep. We performed polysomnography. The apnea/hypopnea index was 43.8/hr, and the mean oxygen saturation during sleep was 76.8% (Fig. 4). We diagnosed sleep apnea and pulmonary hypertension based on the above results and we started performing nasal continuous positive airway pressure. His general condition was improved and the peak velocity of tricuspid regurgitation decreased to 2.7 m/sec. After 6 months, we were able to stop the above mentioned management for sleep apnea. A follow-up of polysomnography was skipped due to improved clinical status. At the last follow-up at the age of 1.5 years, he was doing well without taking any medication except nasal continuous positive airway pressure.
Discussion
OSA can induce systemic hypertension and pulmonary hypertension6-8). Cases of systemic hypertension from OSA were reported in individuals with RTS4,5). However, we could not find any reports of pulmonary hypertension due to sleep apnea in children with RTS.
RTS is a rare syndrome with a frequency of 1 in 100,000 to 125,000 newborns9). The syndrome is characterized by mental retardation, growth retardation and a particular dysmorphology. The characteristic craniofacial features are downslanting palpebral fissures, a columella extending below the nares, a highly arched palate, a grimacing smile and talon cusps. Other variable findings are obesity, coloboma, cataract, congenital heart defects, renal abnormalities and cryptorchidism. Approximately one-third of the affected individuals have a variety of congenital heart diseases. Therefore, all these children should receive an evaluation by a pediatric cardiologist.
Pulmonary hypertension can be induced by a large PDA and other types of congenital heart disease in RTS2,3). These patients can also have upper airway obstruction during sleep due to hypotonia, obesity and the susceptible anatomy of the oropharynx and airway (small nasal passages, retrognathia, micrognathia, hypertrophy of the tonsils and adenoids)2-5). Repetitive episodes of upper airway narrowing and intermittent hypoxia due to sleep related events in OSA lead to vasoconstriction throughout activation of the rennin-angiotensin system and the increase of the level of endothelin-1. Furthermore, intermittent hypoxic events result in oxidative stress, as evidenced by elevated levels of xanthine oxidoreductase and lipid peroxidation. These effects are cumulative over time, potentially forming the basis for elevated cardiovascular risk in individuals with OSA10,11).
Additionally, hypoxia is a causative factor of pulmonary hypertension10-12). In 1947, Motley et al.13) demonstrated that breathing a low oxygen gas (10% O2) induced a rise in pulmonary arterial pressure due to pulmonary vasoconstriction. This pulmonary vasoconstriction induced by repeated hypoxia may cause pulmonary vascular remodeling, which may or may not be reversible, potentially contributing to the development of pulmonary hypertension, as proved in populations with advanced lung disorders like chronic obstructive pulmonary disease10-12).
We experienced a case of a patient with RTS and pulmonary hypertension, and the latter was due to congenital heart defects and OSA successively during infancy. A pulmonary hypertension recurred after the repair of cardiac anomalies, and improved after the control of OSA in this patient. So, inspection of the respiratory pattern and percutaneous oxygen saturation during sleep are needed for patients with RTS and pulmonary hypertension, and performing polysomnography should be considered for confirming the presence of pulmonary hypertension due to obstructive sleep apnea.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


