

Introduction
Pelizaeus-Merzbacher disease (PMD) is a rare, X-linked recessive dysmyelinating disorder of the central nervous system, clinically characterized by nystagmus, spasticity, ataxia, and mental retardation1,2). The estimated incidence is 1 case per 77,000 live births in Germany1). PMD is caused by mutations in the gene encoding PLP1, a myelin protein that is expressed in oligodendrocytes3). Although defects in the proteolipid protein (PLP) gene cause PMD, analysis of the PLP gene has failed to reveal mutations in approximately 10 to 20% of these patients1). Brain magnetic resonance (MR) imaging and MR spectroscopy in PMD patients typically show a lack of myelination in the white matter and specific changes in metabolites, respectively4-6). Proton MR spectroscopy is regarded as a good method to clarify perturbation of brain metabolism7,8). Here, we present a series of 5 cases of PMD, in which the diagnostic value of proton MR spectroscopy was evaluated, in combination with clinical manifestations, brain MR images, and genetic analyses.
Case Report
1. Case 1
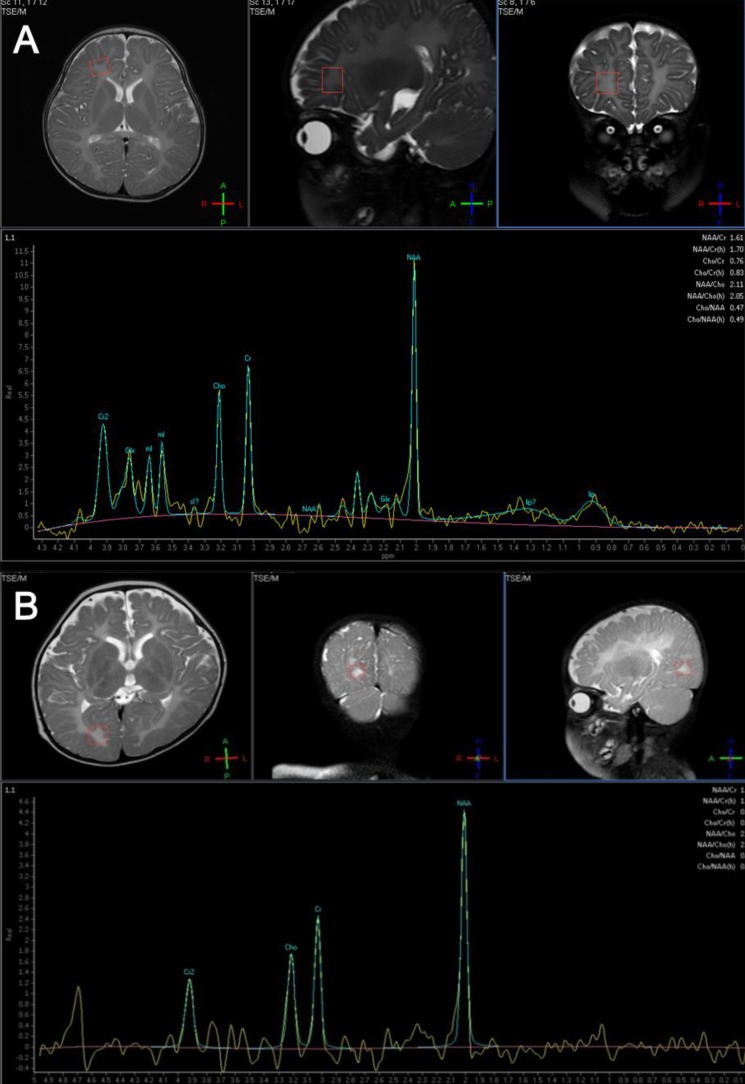
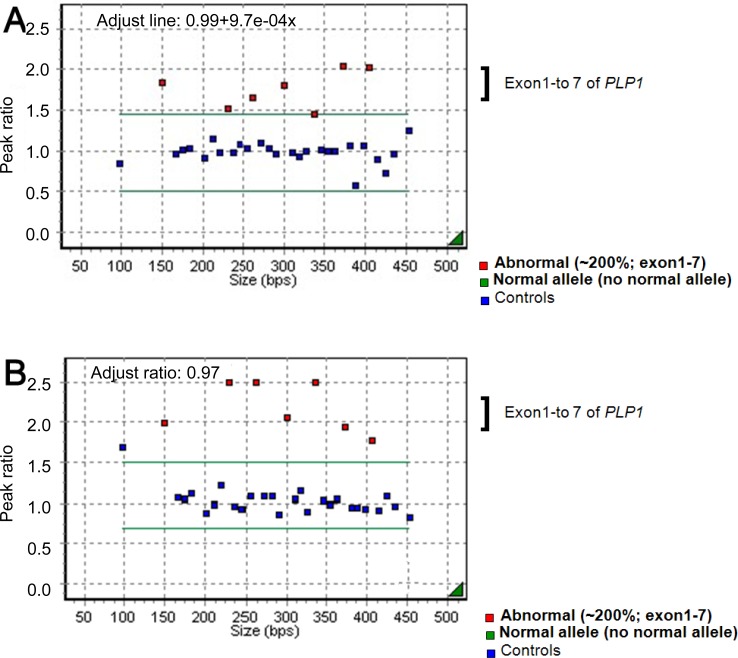
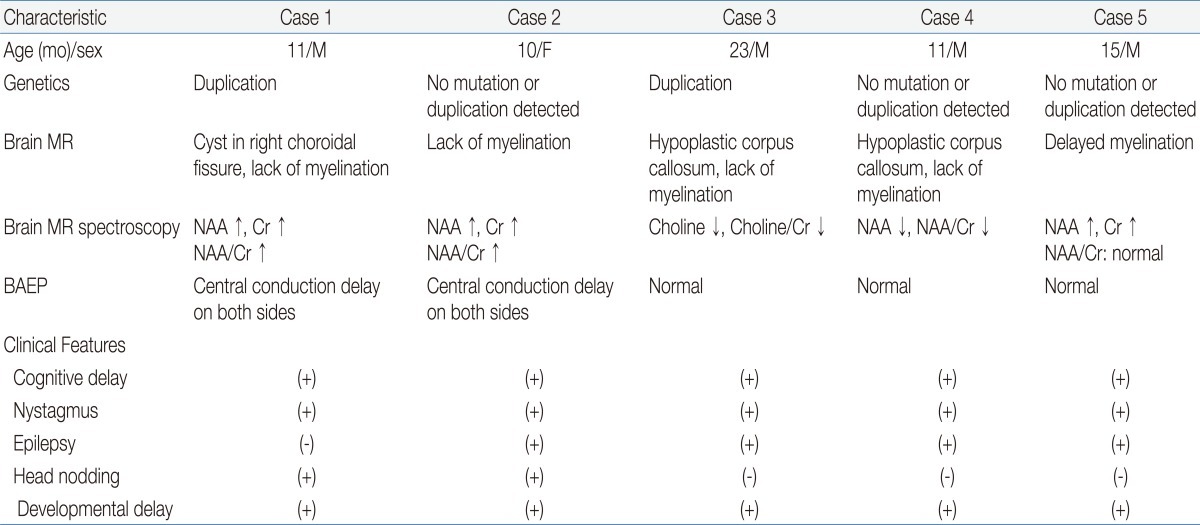
An 11-month-old boy was admitted to Asan Medical Center for evaluation of developmental delays. He was born at term, after a normal pregnancy, with a birth weight of 2,600 g and normal Apgar scores. He had a maternal uncle with developmental delay, but no other family history of related diseases. The patient presented with nystagmus, spasticity, strabismus, and head nodding. When he was 1 month old, his parents noticed jerky eye movements and nodding of his head. Thereafter, he showed frequent head nodding and mild hypertonia in the lower extremities. At the age of 7 months, he underwent a hearing evaluation at another hospital because of suspected deafness. Brainstem auditory evoked potentials were normal, and brain MR imaging showed a choroidal cyst in the right medial temporal lobe. By 11 months of age, he was still unable to sit unaided. At this point, laboratory tests, including those to assess the serum amino acid and urine organic acid levels, were normal. Brain MR imaging was performed and revealed the absence of myelination of the white matter; there had been no progression of myelination compared to brain MR images obtained at 7 months of age (Fig. 1A). MR spectroscopy showed increased NAA and creatine peaks in the white matter and basal ganglia (Fig. 2A). Genetic analysis by multiplex ligation-dependent probe amplification (MLPA) showed duplication of the PLP1 gene (Fig. 3A).
2. Case 2
A 9-month-old girl was referred to Asan Medical Center for evaluation of developmental delays and microcephaly. She was born at term, after a normal pregnancy, with a birth weight of 2,210 g. She did not experience any significant perinatal distress. She was the first child of healthy parents and had no family history of developmental delays. Her height was normal for her age, but her head circumference was below the 3rd percentile for children of her age. A neurologic examination revealed spasticity in all extremities and tremors in the upper extremities. At 1 month of age, fast horizontal nystagmus was noticed. She had difficulty feeding and was unable to hold her head up until she was 8 months old. Even at 9 months of age, she could not maintain a sitting position, and laboratory tests, including serum amino acids and urine organic acid levels, showed normal results. At this point, her developmental state was equivalent to that of a 3- to 4-month-old infant, by the Korean infant and child developmental screening test (KICDT). Electroencephalography (EEG) showed occasional high amplitude delta slowings over both posterior head regions. Analysis of brainstem auditory evoked potentials revealed a central conduction defect on both sides, while visual evoked responses were normal. Brain MR imaging (Fig. 1B) revealed decreased white matter volume and reduced myelin formation, and MR spectroscopy imaging showed increased NAA and creatine levels in the basal ganglia, internal capsule, frontal white matter, and occipital white matter (Fig. 2B). Genetic analysis of the PLP1 gene by MLPA showed no duplication or deletion.
3. Case 3
A 23-month-old boy with developmental delay was admitted for evaluation of tonic contraction. He was the second child of healthy parents, and his 6-year-old brother was also healthy. He was born at full term and did not experience any abnormal perinatal events. At 4 months of age, he experienced spastic contractures of the lower limbs and was diagnosed with developmental delays at another hospital. By the age of 23 months, he was unable to support his head or turn from a prone to supine position. He had feeding problems, with recurrent vomiting. At 23 months of age, his head circumference was 46 cm (P<3 percentile), length was 85.2 cm (P<3 percentile), and weight was 8.6 kg (P<3 percentile). Neurologic examinations revealed nystagmus in all directions, increased muscle tone, and a positive Babinski sign. Laboratory tests of blood and cerebrospinal fluid yielded normal results. A video fluoroscopic swallowing study revealed swallowing dysfunction and silent aspiration that eventually led to the need for a gastrostomy. Brain MR imaging (Fig. 1C) showed delayed myelination, and MR spectroscopy revealed a decreased choline peak in the white matter. An EEG showed nonspecific diffuse cerebral dysfunction. Genetic analyses revealed duplication of the PLP1 gene (Fig. 3B). Subsequently, the patient experienced progressive contracture and prominent spasticity and was on the medication of phenobarbital due to seizures.
4. Case 4
An 11-months-old boy was admitted for treatment of seizures, which began on the day after his birth. He was born after a full-term, unremarkable pregnancy and delivery, with a birth weight of 3,400 g. On admission, his body weight, height, and head circumference were normal for his age. The patient presented with spastic paraplegia, gastroesophageal reflux, dysphagia, nystagmus, and developmental delays. Auditory evoked potentials were normal, as were laboratory results for serum amino acids and urine organic acids. Genetic analysis of the PLP1 gene revealed no duplication. EEGs performed at 5 months and 9 months of age showed multifocal spikes in the frontotemporal regions and hypsarrhythmia, respectively. Brain MR images taken when the patient was 1 year old showed evidence of a hypoplastic corpus callosum and a lack of white matter myelination (Fig. 1D). MR spectroscopy demonstrated decreased NAA levels in the left frontal lobe.
5. Case 5
A 15-month-old boy was admitted for evaluation of developmental delay. He weighed 3,900 g at birth after an uneventful pregnancy and term delivery. His parents and older sibling were healthy. At 15 months of age, his growth was normal for his age, but he could not hold his head up and was unable to roll over or crawl. Neurologic examination revealed spasticity in all extremities. An EEG revealed multifocal spike discharges. Brainstem auditory evoked potentials were normal. However, brain MR imaging showed delayed myelination (Fig. 1E) and MR spectroscopy showed increased levels of NAA and creatine in the white matter. Genetic analysis of the PLP1 gene showed no duplications or mutations. At 16 months of age, his developmental level was equivalent to that of a 1- to 3-month-old infant, by the KICDT.
Discussion
Among the numerous leukodystrophies with early onset and no biochemical markers, PMD is one that can be identified through a combination of stringent clinical criteria and demonstration of the abnormal formation of myelin. Abnormal myelination can be demonstrated through electrophysiological studies and brain MR imaging4-6). In this study, PMD was suspected on the basis of the patients' clinical manifestations such as developmental delays, nystagmus, and abnormal muscle tone. Brain MR imaging, MR spectroscopy, and PLP1 genetic analyses were performed to confirm the PMD diagnoses (Table 1).
In contrast to other demyelinating leukodystrophies such as metachromatic leukodystrophy and adrenoleukodystrophy, in which myelin is formed but subsequently destroyed, PMD is characterized by the failure of myelin formation. PMD is caused by mutations in the PLP1 gene on Xq22, which encodes 2 myelin proteins-PLP1 and DM201). The most common mutation is gene duplication, accounting for 60 to 70% of the mutations, followed by missense mutations, insertions, and deletions9). Coding sequence mutations tend to be recessive, but some mutations are more frequently expressed in women. The mechanism leading to the higher incidence of affected women most likely involves random X-inactivation and altered oligodendrocyte differentiation and survival that depends on the severity of the PLP1 mutation9). In the patients described in this report, molecular analysis of the PLP1 gene was conducted using MLPA, which is more accurate than fluorescence in situ hybridization or quantitative polymerase chain reaction for determining PLP1 copy numbers10). Duplication of the PLP1 gene was detected in patients 1 and 3, both of whom were boys. In patient 2, a girl, neither duplication nor deletion of PLP1 was detected.
The clinical features of PMD are variable and include nystagmus, psychomotor delay, seizure, stridor, feeding difficulties, ataxia, and hypotonia progressing to spasticity. Moreover, the degree of dysmyelination is correlated with the severity of the clinical manifestations4,9,11).
Depending on the age of clinical onset, pattern of transmission, and severity of the symptoms at presentation, PMD is classified into 3 subtypes: connatal, classical, and transitional1,2). The connatal form presents in the neonatal period and is more severe; the pattern of inheritance is probably autosomal recessive. The classical form presents during the first few months of life, progresses slowly, and is X-linked. The transitional form has characteristics that are intermediate between the classical and connatal types; this form resembles the connatal type but progresses more slowly. In the present report, all the patients showed cognitive delay, nystagmus, spasticity, and developmental delays (Table 1). In 4 cases, the patients needed anti-epileptic drugs because of seizures, with epileptiform discharges on EEG. Two patients showed gastroesophageal reflux, and one of them required fundoplication surgery. Four patients experienced swallowing or chewing difficulty during their follow-up.
Brain MR images of PMD patients showed diffuse or patchy T2-hyperintensity due to the lack of myelination4-6). In addition, atrophy and decreased white matter volume are often apparent4). The common pathological characteristic of PMD is the lack of myelin sheaths in large areas of white matter, more prominent in the lateral periventricular regions than in the subcortical regions4).
Proton MR spectroscopy reveals changes in cellular metabolism in the central nervous system and may therefore contribute to both the diagnosis of PMD and a better understanding of its pathogenesis4,9). Specifically, changes in NAA concentrations reflect alterations in axonal metabolism, with elevated levels of NAA being associated with myelin loss in the leukodystrophies12,13). To date, only a few cases of PMD have been reported wherein proton MR spectroscopy has contributed to the diagnoses, and they have shown varying results. However, in Korea, there have been several PMD case reports since 1995, none of which involved MR spectroscopy in making the diagnosis14-17).
In studies of patients with PLP1 duplications, MR spectroscopy has revealed increased NAA concentrations in some cases and decreased NAA concentrations in others4). These findings suggest different states of axonal involvement that may be the result of different mutations, different degrees of axonal involvement, and/or different stages of the disease9,18). Changes in the choline levels in brain MR spectroscopy are also associated with the degree of myelination19). In PMD patients, a decreased level of choline is known to be associated with hypomyelination20). In both cases 1 and 2, brain MR spectroscopy showed increased NAA concentrations, despite differences in the copy number of the PLP1 gene. In case 3, MR spectroscopy showed a decreased level of choline that accompanied the hypomyelination. In case 5, a diagnosis of PMD was made on the basis of the clinical features and the increased NAA and creatine concentrations observed by MR spectroscopy, although mutations in the PLP1 gene were not detected. These observations indicate that further investigations are required to more accurately determine the relationship between changes in NAA concentrations and the type of mutation in the PLP1 gene.
On the basis of clinical manifestations, genetic analyses, brain MR imaging, and MR spectroscopic findings, 5 patients (1 girl and 4 boys) were diagnosed with PMD. These case reports suggest that MR spectroscopic images are potentially valuable in evaluating the degree of axonal integrity and myelination in PMD patients. Moreover, we suggest that MR spectroscopy is an important tool for understanding the pathophysiology of PMD. Due to the failure to detect PLP1 gene mutations in 10 to 20% of PMD patients1) and the lack of diagnostic biochemical tests, comprehensive evaluations of patients suspected of having PMD, appear to be helpful in diagnosing PMD and in understanding the disease pathophysiology.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


