


Introduction
Legg-Calve-Perthes (LCP) is the avascular necrosis or idiopathic osteonecrosis of the epiphysis of the femur head1). The main factor that plays a role in the etiology is the decreased blood flow to the epiphysis of the femur head. It is more frequently observed among male children between the ages of two and 12 years1,2).
Hypopituitarism is a clinical syndrome that develops due to a partial or complete insufficiency of all, or one or more, of the hormones produced in the pituitary gland3). Growth hormone deficiency (GHD) is typically characterized by decreased growth hormone (GH) secretion despite spontaneous and pharmacological warnings when there are no other causes to explain significant growth retardation, slow growth rate, bone age lag, and short stature. Causes of GHD include genetic (e.g., GH or GH-secreting hormone gene defects), anatomical or congenital (e.g., midline defects, septo-optic dysplasia, or vascular malformations), and acquired (e.g., craniopharyngioma, glioma, or histiocytosis) hypothalamic and hypophyseal anomalies4,5).
The current article presents a patient with complaints of short stature, a diagnosis of multiple hypophyseal hormone deficiency, and a history of LCP and difficult birth-related pituitary stalk interruption syndrome. The case was presented for the following reasons: LCP and hypophyseal hormone deficiency might have developed secondary to difficult birth; LCP might have developed secondary to insulin-like growth factor (IGF)-1 deficiency related to GH deficiency; and this association has not been previously published.
Case report
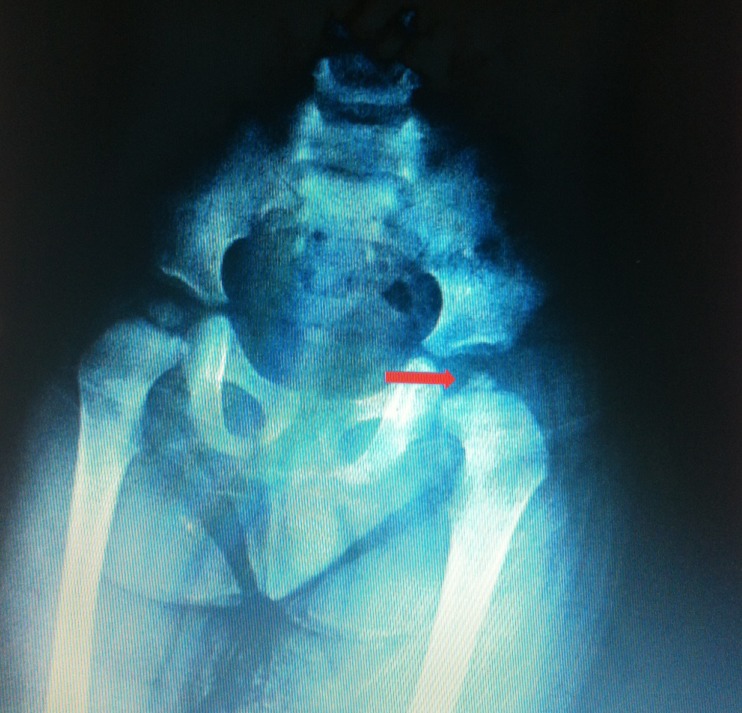
A male patient aged four years and 10 months was brought in with a complaint of short stature. It was indicated that although his development was normal until 12 months of age, it halted afterwards. It was found out that the patient was born at term in breech position through cesarean section with a normal Apgar score and birth weight of 3,750 g, hospitalized postnatally for five days due to birth in breech position and was not noted to be hypothermic and hypoglycemic, was diagnosed with LCP at four years of age following detailed examination due to complaints of pain in the hips, and consequently underwent surgery (Fig. 1). Family history indicated that the patient's parents were related to each other within the second degree of consanguinity. His first sibling was being followed up for Tangier disease, and his second sibling was being followed up for hyperlipidemia by the Department of Pediatric Metabolism. Physical examination findings showed that the patient was prepubertal, and that he had a height of 96 cm (standard deviation score [SDS], -3.2), body weight of 14.6 kg (SDS, -1.9), 2 cm shorter left leg compared to right leg, a surgical scar in the left inguinal region, and stretched penile length of 4.5 cm. Other systemic examination findings were normal. While the patient's chronological age was four years and 10 months, his bone age was consistent with a 2-year-old's (according to Greulich and Pyle).
Laboratory tests yielded the following results: white blood cell count, 11,300/mm3; hemoglobin, 12.1 g/dL; thrombocyte, 320,000/mm3; urea, 7 mg/dL; creatinine, 0.9 ml/dL; serum sodium, 148 mmol/L; potassium, 4.3 mmol/L; chloride, 102 mmol/L; calcium, 9.7 mg/dL; phosphorus, 4.4 mg/dL; normal liver function; fasting blood glucose, 92 mg/dL; total cholesterol, 130 mg/dL; high-density lipoprotein, 41 mg/dL; low density lipoprotein cholesterol, 69 mg/dL; triglyceride, 50 mg/dL; and urine density, 1,022 g/cm3.
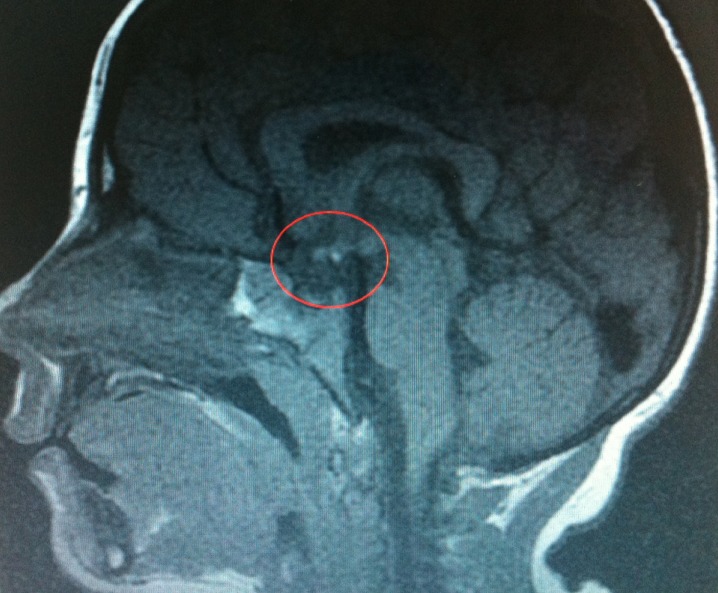
Detailed laboratory analyses showed thyroid function consistent with central hypothyroidism (sT4, 0.49 ng/dL [0.8-1.9 ng/dL]; thyroid-stimulating hormone, 2.57 mU/L [0.5-5 mU/L]). The patient was also determined to have hypocortisolemia (basal serum cortisol level of 2.75 g/dL, stimulated serum cortisol level of 5.71 g/dL [with low-dose adrenocorticotropic hormone]) and GHD (peak GH values in glucagon and levodopa tests while the patient was euthyroid were 0.257 and 0.46 ng/mL, respectively). Serum IGF-1 level was 15 ng/mL (SDS, -3) and Insulin-like growth factor binding protein-3 (IGFBP3) level was 900 ng/mL (SDS, -2.6). An etiology-oriented hypophyseal magnetic resonance imaging was performed due to high prolactin levels (37.4 ng/mL [normal range, 0-25 ng/mL]), which revealed in the sellar cavity center that the adenohypophyseal gland height was 3 mm, neurohypophyseal localization was abnormal, and that there was no signal intensity. In addition, T1A hyperintense nodular appearance with dimensions of 3 mm×2.5 mm showing diffused homogeneous contrast retention following injection of contrast agent was observed at the median eminence level (ectopic neurohypophysis). The pituitary stalk was not identified as a separate structure (Fig. 2). Considering the patient's medical history, it was deduced and evaluated as a pituitary stalk interruption syndrome due to difficult birth. The patient was sequentially started on hydrocortisone, L-thyroxin, and GH therapy.
Discussion
Although several causes have been believed to be responsible for the development of avascular necrosis of the femur head, none of them, except trauma, have yet to completely explain the biological process that plays a role in the development of this pathological phenomenon. It is known that osteonecrosis can develop as a result of trauma even in the absence of a fracture1,2). Although IGF-1 levels have been found to be lower in LCP patients compared to normal population in the literature6), the present case with a complaint of short stature, a diagnosis of multiple hypophyseal hormone deficiency, and a history of LCP, was presented for the following reasons: LCP and hypophyseal hormone deficiency might have developed secondary to difficult birth; LCP might have developed secondary to IGF-1 deficiency related to GH deficiency; and this association has not been previously published.
LCP is most frequently observed in males (male:female, 5:1) aged between two and 12 years7). Although increased rates of LCP incidence have been reported in the presence of various factors, such as low birth weight, low socioeconomic development, and exposure to cigarette smoke, the main factor that plays a role in the etiology of the disease is decreased blood flow to the epiphysis of the femur head1,2). Furthermore, many predisposing factors have been suggested for the etiology of the disease, most showing varying degrees of effects in every study1,2,6,7). Diagnosis is based on clinical and radiological findings1,2). The present case was a male within the age group that the disease is most frequently observed in. He was at normal birth weight for the gestational month, and had no prenatal exposure to cigarette smoke. He also had no history of trauma other than at birth, and his diagnosis was made based on clinical and radiological findings.
GH is primarily effective in bone and muscle tissues. It stimulates the synthesis and secretion of IGF-1 in the liver, and the autocrine and paracrine effects of IGF-1 increase its production. Postnatal growth of cartilage tissue depends on hypophyseal GH, and IGF-1 partly mediates this effect4,5,8). While the primary agent is IGF-1 in the postnatal growth of cartilage, it is IGF-2 during the prenatal period. IGF-1 is not only produced in the liver and kidneys, but also in the chondrocytes of the growing cartilage. IGF-1, produced in the chondrocytes, stimulates the growth of cartilage by binding to the IGF-1 receptors in the chondrocytes with its autocrine effects4,8). IGF-1 has a direct effect on the differentiation of prechondrocytes to young chondrocytes and their clonal growth. This, consequently, results in growth4,5,8).
Studies in the literature showed that mice with excised hypophysis exhibited very little growth when IGF-1 was administered locally to the bone epiphysis. This is because differentiated cells required for the binding of IGF-1 appear only after the differentiation of precursor cells by GH in the germinal layer of the growth plate. In studies conducted on mice with excised hypophysis, both administering GH alone and IGF-1 alone led to an increase in body weight, long bone growth, and cell division9,10). Following administration, GH stimulates IGF-1 production in tissues, and consequently, growth through its autocrine and paracrine effects. When IGF-1 is administered, it travels directly to the target tissues and exerts mitogenic effects10,11). Many researchers have agreed that abnormal IGF-1 levels have an effect on the pathogenesis of the disease in LCP patients6,9,10,11).
The present case exhibited significant short stature due to congenital GH deficiency. We suggest that GH deficiency, and IGF-1 deficiency secondary to GH deficiency, caused the short stature and osteonecrosis in the current case by affecting bone development through the abovementioned mechanisms.
In the literature, even though IGF-1 levels were determined to be lower in LCP patients than in normal population, there was no difference in IGFBP3 levels. There was also no difference in hypophyseal hormone levels between LCP patients who underwent surgery and those who have not6,12). In a different study where anterior hypophyseal hormones were evaluated in 139 pediatric LCP patients, hypophyseal hormone deficiency was not observed in any of the patients13).
In conclusion, even though IGF-1 levels have been determined to be lower in LCP patients than in normal population in the literature, the present case study was in which LCP was identified in a patient with GHD as a part of multiple hypophyseal hormone deficiency. We propose that IGF-1 deficiency secondary to GH deficiency can be an effective factor in the etiology of LCP, and that patients with GH deficiency should be monitored closely for LCP development both before treatment and during GH therapy.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


