


Introduction
Interstitial deletions of chromosome 15q outside the deletions at 15q11q13, which result in either Prader-Willi syndrome or Angelman syndrome, are relatively rare1,2). Ring chromosome 15 syndrome and distal 15q deletions are far more frequent reported than interstitial 15 deletions3). To the best of our knowledge only eleven cases with an interstitial deletion of chromosome 15, other than 15q11q13 region, have been reported2,4,5). Interstitial deletions involving the chromosome band 15q22q24 are very rare and only nine cases were previously reported. Although the phenotype of the nine cases is variable and depends on the extent of deletion, all affected patients are reported to have significant developmental delays and most have altered muscle tone (typically hypotonia). In addition, eye abnormalities (strabismus, microphthalmia, colobomas), ear abnormalities (cleft ear lobe, preauricular tags), and urologic defects are also common feature6,7). In this report, we describe a patient with interstitial deletions involving 15q22q23 with global developmental delay, hypotonia, and sensory neural hearing loss. This is the first case in Korea.
Case report
A 12-day-old boy was referred to Dong-A University Hospital due to failure of hearing screening test. He was born by elective cesarean section with a birth weight 3,120 g at 41+3-week gestation. Apgar score was 8 at one minute and 9 at five minutes, and there were no perinatal problems. He was the first child of a healthy 34-year-old mother and 35-year-old father. There were no problems on the antenatal care. There was no history of mental retardation or congenital anomaly in the family. On physical examination at admission, his weight was 3,100 g (10th-50th percentile), length was 49 cm (10th-50th percentile), head circumference was 35 cm (10th-50th percentile). He was noted to have hypotonia with head lagging. There was a frontal bossing, flat nasal bridge, microretrognathia with normal palate and uvula, thin upper lip with inverted V-shape, a midline sacral dimple, and severe both calcanovalgus. His sucking power was weak, he was fed with orogastric tube during 7 days after admission. Brain magnetic resonance imaging (MRI), echocardiogram, abdominal sonogram, spine x-ray, spine sonogram, and ophthalmologic examination were normal. The auditory brainstem response threshold was 50 dB on both side. He was suggested to have sensory neural hearing loss. At 3 months of age and 6 months of age, he was re-evaluated for sensory neural hearing loss. The auditory brainstem response threshold was 40 dB for the right ear and 50 dB for the left ear. Use of a hearing aid and speech evaluation started at 6 months.
On physical examination at 18 months of age, he had severe failure to thrive. His body weight was 8.0 kg (<1st percentile), length was 79.4 cm (17th percentile), and head circumference was 46.1 cm (15th percentile) according to the Korean standards. He had severe global developmental delay and hypotonia. His cognitive composite score was 55 (0.1th percentile), language composite score was 47 (<0.1th percentile), and motor composite score was 46 (<0.1th percentile) at the Bayley developmental test III. His developmental age was around 3 months 10 days at cognition, <16 days at receptive communication, 20 days at expressive communication, 3 months 10 days at fine motor, 2 months 10 days at gross motor according to the Bayley developmental test III.
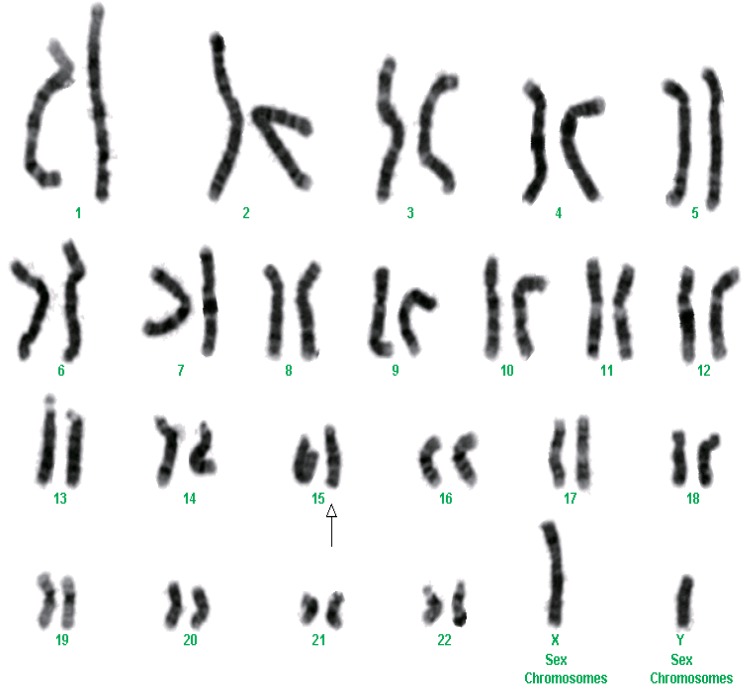
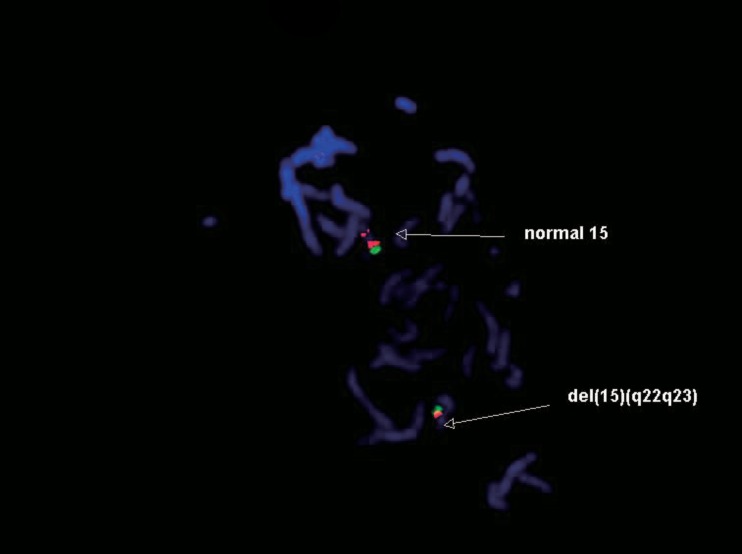
After we obtained informed consents, blood samples were collected from the patient at 12-day age and his parents for chromosome analysis. Chromosome analyses from peripheral blood lymphocytes were performed by traditional synchronization methods and G-banding. One of the chromosomes 15 showed an interstitial deletion in the midportion of the long arm in all 23 metaphase cells examined. Fluorescence in situ hybridization (FISH) studies using Prader-Willi/Angelman region probes (Vysis Inc., Downers Grove, IL, USA) detected deletions of PML gene located in 15q22, leaving both D15Z1 and SNRPN gene signals intact. The latter ones locate in 15p11.2 and 15q11q13, respectively. These FISH findings confirm that the deleted regions contain at least 15q22. The chromosome analysis was written as 46, XY,del(15)(q22q23) (Figs. 1, 2). Parental chromosomes were performed and were normal.
Discussion
In this report, we describe a patient with interstitial deletion involving 15q22q23. The clinical manifestations included marked global developmental delay, hypotonia, sensory neural hearing loss, frontal bossing, flat nasal bridge, microretrognathia with normal palate and uvula, thin upper lip with inverted V-shape, a midline sacral dimple, and severe both calcanovalgus.
Smith et al.8) reported a single patient with submicroscopic deletions at 15q22q23. The patient, a 7-year-old female, reportedly demonstrated features similar to our case, including hypotonia and global developmental delay. She had a long, narrow face, slight flat occiput with no horizontal groove, minimal epicanthal folds, slightly concave nasal base and bridge, thin upper lip. The patient was also diagnosed with autism at age of 4 years, a feature not yet described in other patients but which may be of significant prognostic value. The authors utilized multiple probes that mapped to the region to delineate breakpoints. In their report, the PML and D15S124 probes, localized to 15q22q23, failed to hybridize to one copy of chromosome 15. Furthermore, the neighboring probes showed marked reduced hybridization. Therefore, they concluded that the deletion was approximately 1 megabase in size and encompassed 15q22q238).
Cushman et al.9) reported a patient with an interstitial deletion of chromosome 15q22.3q24. The 15-day-old boy had mild hypotonia, flat nasal bridge, moderate micrognathia with normal palate and uvula, notched earlobe, a sacral dimple with extra bony segment on sacral ultrasonography, and mild hypospadias. The cytogenetic nomenclature was written as: 46, XY, del(15) (q22.3q24). ish del(15)(D15Z1x2, SNRPNx2, PML-). Parental G-banded chromosomes and FISH analyses were normal9).
Formiga et al.10) reported two cases with slightly different karyotypes. The first female patient had a deletion spanning 15q22q25 with feeding difficulties, growth deficiency, psychomotor retardation with no acquired milestones, clonic seizures of the extremities, epileptic apnea, and moderate cortical atrophy on MRI. She also had hypertelorism, epicanthal folds, narrow, slanting palpebral fissures, microretrognathia, microphalmia, and microcephaly. The patient died at 2 years of age due to severe respiratory illness. The second female patient with a deletion involving 15q21q24 had respiratory distress at neonatal period and feeding difficulties requiring tube feeding. She also had septal hypertrophy with dilation of the aorta and pulmonary artery, an abnormal electroencephalography, axial hypotonia, and psychomotor retardation. A physical examination revealed small, slanting palpebral fissures, moderate microphthalmia, microretrognathia, an arched palate, abnormal insertion of toes, and single palmar crease. The patient died at 8 months of age secondary to respiratory distress. In both cases, parental chromosome were normal10).
Clark6) reported a 10-day-old female with a deletion at 15q22q24. She was found to have cystic renal dysplasia and hypoplasia, pulmonary hypoplasia, a flat face, dislocated knees, ventricular septal hypertrophy and an endocaedial cushion defect with a common atrioventricular valve on echocardiography. The patient died 15 hours after evaluation due to respiratory distress. The mother's chromosome was normal6).
The majority of these cases with deletions involving 15q22q24 share several characteristics, including hypotonia, feeding difficulties, and global developmental delay. They also have similar dysmorphic features including a flat face with flat nasal bridge, epicanthal fold, micrognathia, microphalmia, mild abnormality of skeletal and urogenital system. In contrast to those with larger deletions involving 15q22q24, the patient with cryptic deletions usually manifest less serious anomalies and subtle facial dysmorphisms. Despite cryptic deletion of 15q22q23, our case had severe global developmental delay, severe failure to thrive and sensory neural hearing loss.
In conclusion, we have described a new patient with interstitial deletion of 15q22q23. Clinical manifestations including severe global developmental delays, hypotonia, sensory neural hearing loss, and dysmorphic face are associated with a cytogenic deletion. A FISH analysis with a PML probe should be performed in patients with these physical findings.
On the basis of the clinical features as well as chromosome analysis, the diagnosis of 15q22q23 was confirmed. Our study might help delineate a better genotype-phenotype correlation associated with 15q22q23 deletions
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


