

Introduction
Mitochondria are the main energy producing cellular organelles1). Mitochondrial disease (MD) is an inherited metabolic disorder which generally affects the cellular energy metabolism of tissues due to defects in the mitochondrial respiratory chain complex (MRC)2). These MRC defects affect cellular function in several of ways3). The most commonly recognized clinical manifestations of MRC defects are related to the neuromuscular system. However, MRC defects can affect any organ system in the human body, and the clinical symptoms vary greatly depending on the organs affected4, 5).
Cardiovascular involvement has been studied as an important prognostic factor in many diseases6-10). It follows that cardiovascular involvement could also be an important prognostic factor in MD11, 12). A previous study reported that the prevalence of cardiovascular involvement ranked second behind visceral involvement in patients with MD13). Also, Finsterer14) followed 113 MRC defect patients over a ten-year period and found that 40% of the patients developed heart disease including cardiomyopathy and heart failure. But unfortunately, most of the studies have been made on cardiovascular involvement or cardiomyopathy in children with specific classic mitochondrial syndrome such as mitochondrial encephalopathy, lactic acidosis, and stroke like syndrome (MELAS) or Kearns-sayre syndrome or with specific mitochondrial DNA mutation. Little reports were made concerning cardiac conduction in patients with diverse mitochondrial diseases not to mention in Korean patients.
In this study, we analyzed characteristics of cardiac conductivity using ECG in patients with nonspecific MD who take higher percentage than those with specific types among mitochondrial disease population.
Materials and methods
1. Patients group
We performed a retrospective study on 57 MD patients with MRC defect and nonspecific encephalopathy who did not have clinical cardiovascular disease manifestations. The patients had been previously treated in the Department of Pediatrics of Gangnam Severance Hospital from March 2006 to February 2008. All MRC defect patients were confirmed by biochemical enzyme assays of muscle tissue. We excluded the specific mitochondrial syndrome and only included patients with MRC defect I, the most common type of MRC defect, to increase the homogeneity of the study.
We evaluated mitochondrial enzyme function of isolated mitochondria collected from muscle tissue using the spectrophotometric assay previously described by Rustin et al15). The activities of nicotinamide adenine dinucleotide (NADH), coenzyme Q (CoQ) reductase (complex I), succinate-CoQ reductase (complex II), succinate-cytochrome c reductase (complex II-III), cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), oligomycin-sensitive ATPase (complex V) and citrate synthase were evaluated and we defined MRC defects as residual enzyme activity of less than 20% compared to that of the age-matched controls.
2. Electrocardiography
We performed standard 12-lead ECG on all patients and recorded amplitude by 1 mV/10 mm and speed by 25 mm/sec. All patients were in the supine position during the ECGs. Cardiologists analyzed all ECGs retrospectively from paper printouts. We measured RR and QT intervals in lead II and then corrected QT (QTc) by Bazett's formula, QTc = QT / √RR16).
The abnormalities on the ECGs were compared with the normal reference ranges7, 17, 18) (Normal PR interval: 90 to 170 ms for 7-15 years old and 120 to 200 ms for ≥16 years old; Normal QRS duration: 40 to 90 ms for 7-15 years and 50 to 100 ms for ≥16 years old; Normal QTc: ≤440 ms; Abnormal T wave: QRS-T angle >60° in limb lead, T wave <10% of QRS height or negative T wave in leads V4-6).
Results
1. Patient characteristics
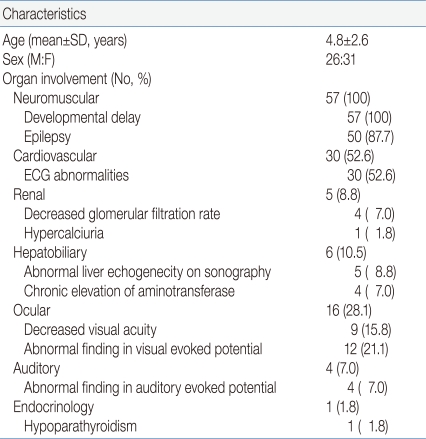
The mean age of MRC defect patients was 4.8±2.6 (mean±SD, years) and the gender ratio was 1:19 (Male:Female). Regarding clinical manifestations, all 57 patients (100%) showed neuromuscular system involvement, all showed delayed development and 50 (87.7%) suffered from epilepsy. Cardiovascular involvement was detected in 30 patients (52.6%) by ECG results. There were five patients (8.8%) with renal system involvement, of which four (7.0%) had decreased glomerular filtration rate and one (1.8%) had hypercalciuria (random urine Calcium/Creatinine ratio >0.2). Hepatobiliary involvement was seen in six patients (10.5%). Five (8.8%) patients had abnormal liver echogenicity on sonography and four (7.0%) had chronic elevation of amino transferase. Ophthalmologic involvement was seen in 16 patients (28.1%). Decreased visual acuity was noted in nine (15.8%) children and abnormal visual evoked potential in 12 (21.1%). Auditory involvement in four (7.0%), and endocrine involvement in one (1.8%) with hypoparathyroidism (Table 1).
2. ECG findings
ECG abnormalities were observed in 30 patients (52.6%). Prolongation of the QTc interval (>440 ms) was seen in 19 patients (33.3%), widening of the corrected QRS interval in 15 (26.3%), and bundle branch block in four (7.0%). Atrioventricular block, premature atrial contraction and premature ventricular contraction were seen in two patients each (3.5%). Wolff-Parkinson-White syndrome was noted in one patient (1.8%) (Table 2).
Discussion
In this study, prolongation of QTc was the most frequent pathologic finding on ECG. The QT interval reflects the total duration of ventricular repolarization and depolarization, and the QTc is a known predictor of cardiovascular mortality in patients with and without known cardiac disease18, 19). Prolongation of QTc can cause fatal ventricular arrhythmia as a consequence of early development of depolarization. Many studies have focused on the relationship between QTc prolongation and mortality in a variety of diseases. Salles et al20) studied the relationship between QTc prolongation and mortality risk in patients with type 2 diabetes mellitus and found that the hazard ratio increased among patients with QTc prolongation. Okin et al21) also found that diabetic patients with QTc prolongation had an increased risk of death and a five-year mortality of 25.3% compared with 13.9% in individuals without QTc prolongation. Based on studies that have shown that QTc prolongation is associated with increased mortality, we think that it could be used as an indicator for decreased cardiac function and possible sudden cardiac death in patients with MD.
Previous studies of cardiac involvement in MD patients have mostly focused on cardiomyopathy11-13, 22, 23), and the studies that do focus on conduction abnormalities have been restricted to Kerns-Sayre syndrome, which is associated with long QT syndrome24, 25). In this study, we could observe a variety of ECG abnormalities in our patients. Up until now, the pathophysiology of cardiovascular involvement in MD has been explained by a decrease in adenosine triphosphate (ATP) synthesis and damage by reactive oxygen species. Defects in ATP synthesis seem to affect both cardiac conduction and contraction12, 22).
Our study of nonspecific mitochondrial encephalopathy patients again demonstrated that the heart is the second most frequently involved organ behind the neuromuscular system. Since the heart is heavily dependent on energy metabolism, it is one of the first organs to be affected when there is a defect in MRC11). There have been similar findings in other metabolic disorders. Bonnet et al26) recently found 24 cases of arrhythmias in 107 patients with defects in fatty acid oxidation. In those 24 patients, they also detected accumulations of long-chain acylcarnitine in fibroblasts. These accumulations occurred because the long-chain fatty acid transporters could not cross the mitochondrial inner membrane. Furthermore, Koisumi et al27) found that patients with abnormally low levels of serum free carnitine had a higher risk of ventricular hypertrophy and provided evidence that low carnitine levels could induce electromyocardial changes. More extensive studies on mitochondrial metabolism are needed as seems to be intimately related to those results.
There are some limitations in this study. Since MD is rare we could not perform a prospective study. We did, however, evaluate cardiac function in patients with nonspecific mitochondrial encephalopathy and not just with a specific mitochondrial syndrome, an evaluation which had not yet been done. We found that their cardiac conductivity was affected. Because of this finding, we strongly recommend active screening and regular follow-up of patients with MD using ECG even in patients who do not exhibit typical cardiac manifestations. In addition, we think that not only screening test on conductivity but also contractility using echocardiography would be useful as well. These screening techniques will allow for early detection of any abnormalities in conductivity or contractility that could potentially lead to cardiomyopathy and sudden cardiac death. That being said, further study of cardiac involvement in patients with MD is needed to explore methods for reducing the risks of cardiomyopathy and sudden cardiac death.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


