

Approach to management
The aims of juvenile rheumatic arthritis (JRA) treatment are to control pain; to preserve range of motion (ROM), muscle strength, and function; to manage systemic complications; and to facilitate normal nutrition, growth, and physical and psychological development. Thus, the general aim of treatment is remission rather than improvement. Although the major focus of medical therapy for JRA is on the arthritis, other extra-articular complications (e.g., uveitis, serositis, growth retardation, and osteopenia) require consideration1). In general, the treatment program for this disease should be family-centered, community-based, and well coordinated. An ideal approach involves a multidisciplinary team that consists of a pediatric rheumatologist, nurse clinician, social worker, physical therapist, occupational therapist, and psychologist. In addition, consultation with a physiatrist, psychiatrist, orthopedic surgeon, dentist, or nutritionist is often indicated. Moreover, regular ophthalmologic consultation is mandatory for JRA patients. Because this rheumatic disorder is characterized by chronic or recurrent inflammation of the joints and varying systemic manifestations, the child and his/her family should accept the need for long-term treatment and surveillance for effective management of the disease.
1. Nonsteroidal anti-inflammatory drugs (NSAIDs)
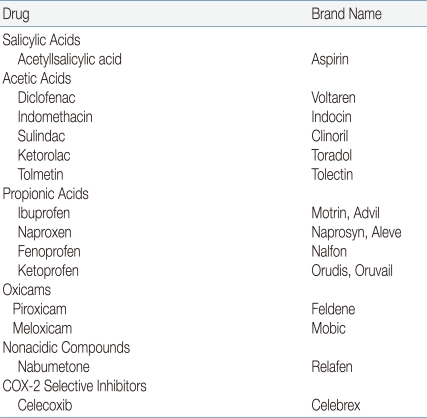
Most children with JRA are treated with one of the NSAIDs during the initial approach of therapy (Table 1). All of these drugs have antipyretic, analgesic, and anti-inflammatory actions, as well as a record of long-term safety. Most currently used NSAIDs inhibit the activity of cyclooxygenases 1 and 2 (COX-1 and COX-2, respectively). Therefore, these drugs have the potential to induce gastrointestinal (GI) irritation2), although this side effect is unusual in children, and ranitidine may ameliorate it. New selective COX-2 inhibitors are available, but have not generally been evaluated in children3). Recent observations in clinical trials involving older adults have suggested an increased risk of cardiovascular events secondary to nonsteroidal anti-inflammatory drugs (NSAIDs), but there are no comparable studies in children. Consideration should be given to continuation of an NSAID for at least 3 to 6 months after all evidence of active disease has disappeared. One might also consider a different mode of withdrawal, such as decreasing administration to every 2 weeks for a period of time before discontinuation1).
Naproxen has been shown effective in the management of joint inflammation in JRA at a dosage of 15 to 20 mg kg-1 d-1 given with food, and it only requires administration twice daily. Naproxen is typically well tolerated, although mild epigastric discomfort has been occasionally encountered. In addition, cutaneous pseudoporphyria is a side effect of this drug, and is characterized by a bullous eruption on the face, hands, or other sun-exposed areas, which often leaves an irregular, shallow scar on the affected individual.
Ibuprofen is a relatively mild anti-inflammatory agent and is typically well tolerated at a dosage of approximately 35 mg kg-1 d-1, divided into 3 or 4 doses given with food. The suspension that is commonly administered to children consists of both S and R enantiomers and is not absorbed as well as the tablets6). It should therefore be given at a dosage of approximately 45 mg kg-1 d-1 divided into 3 or 4 doses.
Tolmetin, which is given with food in 3 divided dosages totaling 25-30 mg kg-1 d-1, is equally effective as an anti-inflammatory drug.
Diclofenac may be useful in children who are unable to tolerable other NSAIDs because of gastric side effects. This drug is typically prescribed in 3 doses per day; a slow-acting preparation is also available.
Sulindac is an inactive prodrug that is converted in vivo to active sulfide, and therefore offers little theoretical exposure to the GI mucosa. It has also been suggested that this prodrug is less nephrotoxic than other NSAIDs.
Celecoxib and, more recently, analogues of the COX-2 inhibitors have been introduced for treatment of arthritis in adults. These drugs are reputedly less likely to cause gastric irritation and peptic ulcer disease than traditional NSAIDs3).
Indomethacin, typically at a dosage of 1-3 mg kg-1 d-1 but up to a maximum of 125 mg d-1, is useful for treating fever and pericarditis associated with systemic disease. In many children, intermittent fever responds only to prednisone or indomethacin, the latter of which is a potent anti-inflammatory drug.
Piroxicam, which is only given once daily, may be particularly useful in older children and adolescents who are sometimes incompliant with taking prescribed medication.
Aspirin was previously the drug of choice in the initial management of inflammation, but has more recently been replaced by the NSAIDs. The reasons for this switch are related more to convenience of administration and relative freedom from side effects than to superior efficacy. In addition, aspirin likely resulted in more frequent instances of transaminasemia than the newer NSAIDs. Aspirin is typically started at 75-90 mg kg-1 d-1 in 4 doses given with food in order to minimize gastric irritation and to ensure therapeutic blood levels. It may be difficult to reach therapeutic levels in children with acute systemic disease, but care should be taken with increasing the dose beyond 130 mg kg-1 because this often results in salicylism. Of note, awakening children at night to administer aspirin is unnecessary because the serum half-life of salicylate is prolonged once therapeutic levels have been achieved. In terms of side effects, aspirin and other NSAIDs are associated with interstitial nephritis and renal papillary necrosis4).
2. Methotrexate
Methotrexate is considered the initial second-line agent for treating most children with chronic arthritis, because of its relatively rapid onset of action, efficacy, and acceptable toxicity. The advantages of this medication are its efficacy at a relatively low dose, oral administration, once-a-week dosing, and apparent lack of oncogenicity and production of sterility9). Most patients respond to this drug by 3 months, although a child may occasionally require a longer period of treatment. Methotrexate therapy should likely be continued for 1 year or longer after remission has been achieved. The principal toxicities of this drug are directed at the bone marrow, liver, and very rarely the lung. However, cirrhosis of the liver is not an expected toxic effect in children on a weekly therapy10), although methotrexate-induced pneumonitis and effects on pulmonary function have been reported in children11). Folic acid, given at 1 mg d-1 during treatment with methotrexate, can reduce GI irritation and mucosal toxicity with no diminution in therapeutic effectiveness. Methotrexate is given as a single weekly dose on an empty stomach with clear liquids 45 minutes before breakfast; the minimum oral starting dose is 10 mg m-2 weekly. If a clinical response is inadequate or if oral administration is associated with nausea or vomiting, a trial of subcutaneous administration of the drug should be attempted. Methotrexate should be discontinued if no objective response is documented or if toxicity develops despite a reduction in dose.
3. Glucocorticoid drugs
Glucocorticoid medications are indicated for uncontrolled or life-threatening systemic disease, the treatment of chronic uveitis, and as an intra-articular agent. Systemic glucocorticoids should be administered to individuals with inflammation only with a well-considered therapeutic plan and a clear set of clinical objectives. Although the use of glucocorticoid drugs alone for suppression of joint inflammation is to be discouraged, low-dose or alternate-day prednisone is of benefit to children with severe polyarthritis that is unresponsive to other therapeutic programs. Moreover, low-dose prednisone can be used as a "bridging" agent in the initial treatment of moderately to severely affected children who are started on other slower-acting, anti-inflammatory drugs at the same time12). For severe uncontrolled systemic manifestations with marked disability, prednisone is often prescribed as a single daily morning dosage of 0.25-1.0 mg kg-1 d-1, or in divided doses for more severe disease. Prolonged use of systemic glucocorticoids has been shown to lead to iatrogenic Cushing's syndrome, growth suppression, fractures, cataracts, and increased susceptibility to overwhelming infection. However, it often becomes difficult to reduce the dose of a glucocorticoid because of a child's adaptation to chronic steroid excess13). Moreover, steroid pseudorheumatism may complicate even slow withdrawal from the drug, particularly at lower dose levels.
Intravenous pulse glucocorticoid therapy offers an alternative approach to serious, unresponsive disease. The effect of this treatment is immediate and it is hoped that long-term toxicity is decreased5). Methylprednisolone is the drug of choice for this therapy, often at a dose of 10-30 mg kg-1 per pulse. Established protocols of this technique consist of single pulses spaced 1 month apart, 3 pulses given sequentially on 3 d each month, or 3 pulses administered on alternate days each month. This therapy should always be given with cardiovascular monitoring of the patient during the infusion and for a time thereafter, paying careful attention to electrolyte and fluid balance, and to the potential for cardiac arrhythmia or acute hypertension.
The protocol for intra-articular glucocorticoid administration is changing, but at the present time it is clearly indicated in the management of oligoarthritis that has not responded to an appropriate program of NSAIDs. Moreover, intra-articular glucocorticoid therapy should be considered in the management of polyarticular disease in which one or several target joints have not responded to NSAIDs or anti-inflammatory drugs. However, intra-articular injections should be given only a limited number of times per patient (3 times in a single joint during 1 year). Triamcinolone hexacetonide has been the drug of choice for large joints at a dose of 20-40 mg. Younger children, in addition to those individuals who are undergoing injection of a hip joint or several joints, may require conscious sedation or light general anesthesia prior to the treatment.
4. Biologic response modifiers
Recent therapeutic approaches for children with unremitting inflammatory disease include soluble TNF-α receptor (TNFR) p75 fusion protein (etanercept) and recombinant monoclonal human immunoglobulin G (IgG) antibody to TNF-α(infliximab and adalimumab). A pivotal trial of adlimumab did prove its efficacy and ultimately resulted in the approval of the Food and Drug Administration (FDA). In addition, anti-interleukin (IL)-1 and anti-IL-6 therapies also look very promising, particularly for systemic disease patients. The costimulation modifier abatacept was also shown to be effective and relatively well tolerated according to a short-term analysis of patients, which also resulted in FDA approval. Continued FDA procedures for monitoring safety will improve the ability to identify short- and long-term toxicities of these new agents6).
Etanercept has become a standard therapy for arthritis that has not responded adequately to methotrexate. It is administered to patients at a minimum dose of 0.4 mg kg-1 subcutaneously twice a week with continuation of previous medications. However, this drug should not be started in any child with an infection or a history of recurrent infections. Furthermore, the risk of reactivation of tuberculosis or the development of granulomatous or fungal disease as a result of this drug must be recognized. The long-term safety profile of etanercept indicated the drug can be maintained up to 8 years of continuous use. Exposure-adjusted rates of serious adverse effects (SAEs) were not shown to increase over time, and the most common new SAEs reported beyond 4 years of drug exposure were a flare or worsening of disease7).
Adalimumab is a fully human, IgG, monoclonal anti-TNF antibody. The preliminary results of a multicenter, randomized, double-blinded, stratified study of this drug in polyarticular course JRA has previously been presented8). Based on the data in this trial, the United States FDA approved adalimumab as treatment to reduce the signs and symptoms of moderately to severely active polyarticular JRA in patients 4 years of age and older, in February 2008 (this was the first such approval for a biologic since 1999, when etanercept was approved)9).
Abatacept is a fully human, soluble fusion protein composed of the extracellular domain of the cytotoxic T lymphocyte-associated antigen cytotoxic T-lymphocyte antigen 4 (CTLA-4) and the Fc component of IgG1, and selectively inhibits the costimulatory signal necessary for full T-cell activation19). This drug was investigated in an international, multicenter, prospective study with a design similar to the ones used in the pivotal etanercept and adalimumab trials in JRA. This trial showed abatacept was effective and generally well tolerated by patients, which ultimately resulted in FDA approval for treatment of polyarticular JRA in March 2008. Although it is difficult to compare results of separate trials, it appears that abatacept is as effective as anti-TNF agents, but its maximal effect may be achieved a few weeks later10).
Infliximab has not been approved for use in children. However, infliximab (either 3 or 6 mg kg-1), in combination with methotrexate, has been shown to produce an important, rapid, and durable clinical effect in children with JRA at 1 year, but the primary efficacy endpoint of this study was not significantly different between the groups given infliximab or placebo at 14 weeks of treatment. Also of note, the lower dose (3 mg kg-1) of infliximab was associated with a substantially higher risk of SAEs, infusion reactions, and the development of antibodies to infliximab, ANAs, and anti-dsDNA compared with the corresponding measures in those individuals given the 6 mg kg-1 dose. Thus, the use of infliximab in children warrants further investigation11).
Recombinant interferon-γ has also been used experimentally. In one study, it was given to 10 children (6 systemic onset, 3 polyarthritis, 1 oligoarthritis), of whom 8 showed significant improvement, and 7 entered remission12).
Anakinra, which is a recombinant human IL-1 receptor antagonist (IL-1Ra), is currently undergoing therapeutic trials for treatment of chronic arthritis in children. There has been great interest in the role of IL-1 inhibition in the treatment of systemic JRA. Anakinra (1 mg kg-1) administered as a subcutaneous injection once daily was previously well tolerated in pediatric patients with JRA. Moreover, infection rates of these patients were low, and no clinically significant abnormalities in laboratory data were observed. However, efficacy results from the previous study were not conclusive because of the small sample size. Pharmacokinetic assessments indicated that a daily dosage of 1 mg kg-1 administered subcutaneously provided adequate exposure for the treatment of JRA. Safety data from this study were consistent with results from larger studies in adults and indicated that anakinra was safe and well tolerated in JRA patients13).
Rilonacept, which is also called IL-1 trap, is a fusion protein composed of human cytokine receptor extracellular domains for both IL-1 type 1 receptor and the IL-1 accessory protein. Preliminary results of a phase II trial involving administration of this drug to systemic JRA patients have been presented and are published in abstract form14); 2 different doses (2.2 and 4.4 mg kg-1) were studied. Marked improvements in fever, rash, and active joint counts were noted at both doses.
Tocilizumab is a monoclonal anti-IL-6 receptor antibody that inhibits IL-6 activity15). It has previously been reported to be effective in treating systemic JRA. Tocilizumab may also be useful for treatment of established amyloidosis.
Rituximab remains to be discussed in the context of JRA in the existing literature. That is, we were unable to find any published studies on rituximab for the treatment of JRA.
Thalidomide has been recommended for treatment of systemic onset arthritis16).
5. Modes of advanced therapy
Slow-acting antirheumatic drugs (SAARDs) or disease-modifying antirheumatic drugs (DMARDs) consist of cyclosporine, sulfasalazine, and antimalarials. Methotrexate has generally replaced the SAARDs in treating advanced cases of JRA. However, SAARDs remain to be considered for children who have an incomplete response to various combinations of an NSAID, methotrexate, and a TNF-α blocker.
Cyclosporine is given at an oral dosage of 3-5 mg kg-1 d-1 in 2 divided doses exactly 12 hours apart. Blood pressure should be determined at home for 2 weeks during treatment with this drug and subsequently evaluated periodically with urinalyses and estimates of renal function. Of note, there may be a small long-term risk of developing lymphoma as a result of treatment. The role of this drug in the treatment of arthritis in children is uncertain, but it is likely critically important in treating reactive hemophagocytosis. Combined therapy of this drug with methotrexate has also been recommended in select children.
Hydroxchloroquine is a useful adjunctive agent for treating chronic arthritis in older children. The therapeutic effect of this drug is usually subtle and is rarely evident before 2 to 3 months of therapy. If this drug causes no improvement in a patient after 6 months of treatment, it should be discontinued. Hydroxychloroquine is never used alone, but is instead added to an NSAID regimen, usually at a dosage of 5-6 mg kg-1 d-1. The medication should be taken with food because it can be a GI irritant. An ophthalmologic examination, including testing of color vision and visual fields, is usually performed before therapy is started and traditionally every 6 months thereafter. Hydroxychloroquine has not been recommended in children younger than 4 years of age, and sometimes in those younger than 7 years of age, because of the inability of young children to discern colors adequately for testing on grids or visual fields.
Sulfasalazine has been reported to have modest efficacy in some children with chronic arthritis, and it has the advantage of a more rapid onset of anti-inflammatory action than that which occurs with other SAARDs. This drug should not be used in children with known hypersensitivity to sulfa drugs or salicylate, impaired renal or hepatic function, or specific contraindications, such as porphyria or glucose-6-phosphate dehydrogenase deficiency. Moreover, severe side effects have been reported in children with systemic onset disease. Sulfasalazine is started at a dosage of 12.5 mg kg-1 d-1 given with food, and this dose is gradually increased until a dosage of 50 mg kg-1 d-1 is reached. The benefits of the drug are usually apparent within 4 to 8 weeks after initiation of therapy.
6. Autologous stem cell transplantation
Bone marrow transplantation has been initiated as an experimental treatment of severe autoimmune diseases including rheumatic diseases unresponsive to conventional therapy. Autologous stem cell transplantation (ASCT) is currently being evaluated, but only in a small number of children. This treatment approach is indicated only for children who are affected by severe active disease that fails to be controlled by conventional strategies, including anti-TNF therapy.
In 1999, Wulffraat and co-workers first reported on ASCT in 4 Dutch children with longstanding and unresponsive JRA. All of these patients responded well and were disease-free during a follow-up period of 6-18 months. Furthermore, bone marrow reconstruction was found to be rapid. To date, approximately 30 patients have undergone ASCT for JRA. Dramatic improvement was reported in 60-65% of the patients following the procedure. Wulffraat reported these results at the American College of Rheumatology 2000 conference.
Conclusion
In summary, there is no treatment algorithm uniformly agreed upon for the management of JRA. In general, NSAIDs can be used in all JRA types to control pain and diminish inflammation. Methotrexate at 15 mg m-2 week-1 should be instituted upon confirmation of the diagnosis of the polyarticular course of JRA. A short course of oral prednisone (0.5-2 mg kg-1) may be required for particularly active forms of disease ). If the response to methotrexate is inadequate, a biologic agent should be added to the drug regimen. However, almost no evidence exists on the various biologic agents, such as etanercept, adalimumab, abatacept, anakinra, infliximab, rituximab, and tocilizumab. Moreover, because of the lack of sound long-term safety data, the existing evidence is insufficient to draw firm conclusions about the balance of the risks and benefits of using any biologic agent for treating JRA. Clinicians should be aware of this lack of evidence when considering biologics for patients with JRA.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


