

Introduction
Gaucher disease (Online Mendelian Inheritance in Man #230800) is the most common autosomal recessive lysosomal storage disorder, which is caused by a β-glucocerebrosidase (GBA) deficiency. Various glucocerebrosidases, members of the sphingolipid family, accumulate in many organs, including the liver, spleen, central nervous system, skeletal system, and lungs1). The GBA gene, which causes Gaucher disease2), is located on chromosome 1q213).
The main presenting signs of Gaucher disease are hepatosplenomegaly, anemia, thrombocytopenia, bone pain, and growth retardation. A bone marrow or liver biopsy can help diagnose Gaucher disease by revealing typical Gaucher cells, macrophages filled with lipid material, but the gold standard is to confirm deficient GBA activity in leukocytes or fibroblasts. Genetic tests can be used as an effective tool for diagnosis as well1). Increased activity of biomarkers, such as acid phosphatase, angiotensin-converting enzyme (ACE), chitotriosidase, and ferritin, reflect disease activity4-6).
Gaucher disease used to be categorized into three subgroups by the Knudsen and Kaplan classification, as types 1, 2, and 3, according to the presence of neurological deterioration, age at identification, and progression rate.
But, currently, it is divided into three different subtypes; non-neuronopathic, acute neuronopathic, and chronic neuronopathic types7-9). The non-neuronopathic type is the most prevalent lysosomal storage disorder as well as the most common Gaucher disease phenotype. Patients are usually identified by splenomegaly in childhood or early adulthood. Hepatomegaly is usually not as severe as splenomegaly. Thrombocytopenia and anemia are easily observed, and skeletal findings such as osteopenia, osteonecrosis, and bone pain are also noted1). Patients with the acute neuronopathic type show extreme abdominal distension due to severe hepatosplenomegaly in infancy. Cranial nerve impairments, such as strabismus, facial muscle weakness, and dysphagia are characteristic, and most patients die within 1 or 2 years of age due to pneumonia or uncontrolled neurological deterioration that consequently leads to respiratory failure10). Patients with chronic neuronopathic type may be identified at any point during childhood by hepatosplenomegaly and or progressive neurological deteriorations such as seizure attack, dementia, hypertonia, and gaze palsy9,11).
More than 350 GBA mutations have been identified to date12). The GBA mutation spectrum varies widely according to ethnic group13). In the Korean population, L444P, G46E, R257Q, and c.630delC are common mutations, but N370S, the most common mutation in Caucasians, has not been found in Korean patients14,15).
Since 1993 when a recombinant GBA was available, enzyme replacement treatment (ERT) has remarkably improved the clinical outcome of Gaucher disease. Particularly, hepatosplenomegaly and hematological abnormalities show notable improvement under ERT in patients with both the non-neuronopathic and chronic neuronopathic types. ERT is now suggested as the standard treatment for non-neuronopathic Gaucher disease16,17). However, because the recombinant enzyme cannot cross the blood-brain barrier, it cannot prevent neurological deterioration in patients with neuronopathic Gaucher disease. Moreover, no curable treatment exists for acute neuronopathic Gaucher disease due to its rapid progression10).
With this background, early detection and treatment are important for patient prognosis. However, the clinical and genetic characteristics of Gaucher disease in the Korean population have not been described in detail. The current study was performed to help pediatricians with the diagnostic process of Gaucher disease as well as to introduce therapeutic strategies tailored to Korean patients.
Materials and methods
The Institutional Review Board of the Asan Medical Center, Seoul, Korea approved this study, and written informed consent was obtained from all subject or parents. Twenty-seven patients diagnosed with Gaucher disease at Asan Medical Center, Seoul, Korea from 1989 to 2011 were included. Clinical findings, including age at diagnosis, presenting symptoms, accompanying signs, laboratory findings at diagnosis, pathological findings from a bone marrow or liver biopsy, GBA activity, GBA mutations, and clinical courses were reviewed for each patient. Patients were divided into three groups: non-neuronopathic, acute neuronopathic, and chronic neuronopathic types. Genomic DNA was extracted from peripheral blood leukocytes using PUREGENE DNA isolation kits (Gentra Systems Inc., Minneapolis, MN, USA). The coding region and flanking intergenic sequences of the GBA gene (GenBank: NG_009783) were sequenced using an ABI3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Biomarkers, including platelet, chitotriosidase, acid phosphatase and ACE, were checked every 3 months since initial diagnostic evaluation.
Results
1. Clinical characteristics of patients with Gaucher disease at diagnosis
Among 20 patients with detailed clinical data available, nine were male, and 11 were female. Eleven patients were classified with the non-neuronopathic type, two patients with the acute neuronopathic type, and the remaining seven patients with the chronic neuronopathic type. Age at diagnosis was 20.8±18.2 years (range, 1.4 to 57 years) in the non-neuronopathic type, and 6.8±5.1 years (range, 2.5 to 15.8 years) in patients with the chronic neuronopathic type. One patient with the acute neuronopathic type was diagnosed at age 7 months due to splenomegaly and hypotonia and died due to pneumonia at age 2 years. The other patient presented with hydrops fetalis, and died at age 40 days. Gaucher disease was diagnosed by postmortem examination (Table 1).
Among 11 patients with the non-neuronopathic type, splenomegaly was found in 10 (90.9%) and thrombocytopenia in 8 (72.7%). Skeletal manifestations such as short stature, scoliosis, and multiple fractures were detected in five cases (45.5%). Another patient (9.1%) presented with B-cell lymphoma but without hepatosplenomegaly. A bone marrow biopsy was performed as a staging work-up, which revealed typical Gaucher cells. In seven patients with the chronic neuronopathic type, splenomegaly (4/7 patients, 57.1%), hepatomegaly (3/7, 42.9%), thrombocytopenia (5/7, 71.4%), and skeletal manifestations (4/7, 57.1%) were also noted, but all these patients had experienced neurological manifestations since the age of 8.7±4.3 years (range, 0.2 to 13.7 years), including epilepsy (4/7, 57.1%), impaired eye movement (3/7, 42.8%), intension tremor (2/7, 28.6%), and cognitive deficits (2/7, 28.6%) (Table 1).
At diagnosis, platelet counts decreased markedly at 72.25±30.06×103/mm3 (range, 16 to 315×103/mm3) in all patients. Hemoglobin level was 12.1±1.7 mg/dL (range, 9.4 to 14.6 mg/dL). Serum aspartate transaminase and alanine transaminase levels were 53.9±50.0 IU/L (range, 11 to 225 IU/L) and 25.9±16.7 IU/L (range, 7 to 58 IU/L), respectively. Plasma chitotriosidase (normal range, 4 to 76 nmol/hr/mL) was markedly increased in 13 of 17 patients (76.5%), but normal in the remaining four (23.5%). The median activity was 3,331.8±2,486.6 nmol/hr/mL (range, 9.4 to 1,5918.1 nmol/hr/mL). ACE increased in 10 of 13 patients (76.9%) with median activity of 145.3±88.2 U/L (range, 28.3 to 324.6 U/L), and acid phosphatase in 13 patients (100%) with median activity of 31.9±18.7 IU/L (range, 11.5 to 73.2 IU/L). These findings were identical in all subtypes.
A histological examination from either the liver (five patients) or bone marrow (15 patients), revealed typical Gaucher cells in 18 of 20 patients. GBA activity decreased at 5.7±3.2 pmol/min/mg (range, 0.2 to 11.6 pmol/min/mg; normal range, 20 to 80 pmol/min/mg) in all patients.
2. Clinical courses of patients with Gaucher disease
The follow-up period was 7.6±5.5 years (range, 0.5 to 17.3 years). Seventeen patients, excluding two with the acute neuronopathic type and one with the chronic neuronopathic type whose parents refused treatment, were treated with recombinant GBA, imiglucerase (Cerezyme, Genzyme, Waltham, MA, USA). ERT (30 to 60 unit/kg q 2 weeks) was started at age 1 7.6±16.0 years (range, 2.1 to 45.0 years), 1.1±0.8 years after diagnosis.
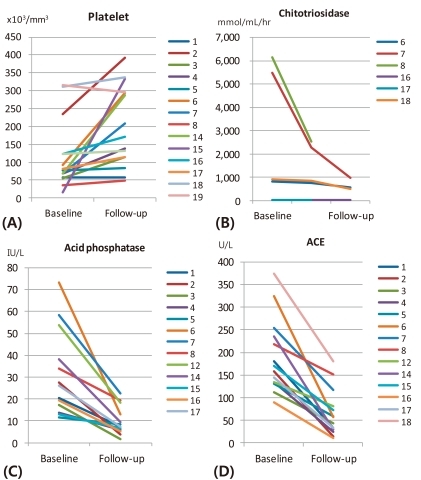
Platelet levels increased to 92.9±75.0×103/mm3 (range, 49 to 393×103/mm3), and normalized in 58.3% of the patients within 6 months of therapy (Fig. 1A). Additionally, chitotriosidase, acid phosphatase, and ACE all decreased to 659.8±280.7 mmol/mL/hr (range, 32.1 to 2,542.9 mmol/mL/hr), 9.6±6.1 IU/L (range, 1.5 to 22.7 IU/L), and 44.6±29.1 U/L (range, 14.2 to 117.3 U/L) within 1 year of therapy, respectively (Fig. 1B-D). A few patients with incomplete data were excluded.
3. GBA mutations in patients with Gaucher disease
A genetic study was conducted in 16 patients. In nine patients with the non-neuronopathic type, L444P (9/18 alleles, 50.0%) and G46E (5/18, 27.8%) were common mutations. Other mutations were D409H (2/18, 11.1%) and R277C (1/18, 5.6%). R277C has not been previously reported. One patient with the acute neuronopathic type, who presented with hydrops fetalis, harbored c.630delC and R257Q mutations. In five patients with the chronic neuronopathic type, L444P was found in four alleles (40%), and N188S, R257Q, and F213I were detected in two alleles (20%) each. All of these have been reported as known mutations in Korean patients with Gaucher disease14).
Discussion
Unlike the non-neuronopathic type that affects Jewish or other Caucasians, the neuronopathic type is relatively more common in East Asians including the Korean population. The acute neuronopathic type was rarely found in our cohort, similar to other ethnic groups18).
Thrombocytopenia was the most prevalent sign, but unlike previous studies18-20), normal liver enzymes or hemoglobin levels were noted in 80.0% (16/20) and 100% (20/20), respectively, of our patients. Skeletal manifestations presented mainly as osteoporosis or short stature in patients with the non-neuronopathic type, whereas variable clinical features were seen in patients with the chronic neuronopathic type, such as frequent fractures or multiple joint involvement.
Chitotriosidase activity, a widely used indicator of treatment response in Gaucher disease, is highly elevated in patients with GBA deficiency, and declines gradually with ERT21), as in our study. However, three patients had normal chitotriosidase activity even before ERT, indicating that they may have functional variants in the chitotriosidase gene, or CHIT122). Previous studies have shown that some patients genetically present with low chitotriosidase activity despite that they are diagnosed with Gaucher disease23), indicating the limitation of chitotriosidase activity as a biomarker to reflect disease activity in some patients. Acid phosphatase and ACE may be used as surrogate biomarkers, as they also decreased with treatment. However, their ranges vary from patient to patient as well.
For 7.6±5.5 years, patients experienced meaningful improvements in quality of life, due to better growth outcomes and a reduction in hepatosplenomegaly. In contrast, none of the neurological symptoms improved during the follow-up period in patients with the chronic neuronopathic type.
The most common mutation in Korean patients with both the non-neuronopathic and neuronopathic types of Gaucher disease is L444P. G46E is a common mutation in patients with the non-neuronopathic type, whereas N188S, F213I, and R257Q occur more frequently in the chronic neuronopathic type. This implies the probability of correlations between their genotypes and phenotypes, which need to be investigated in a larger cohort of Korean patients with Gaucher disease.
Although L444P was a leading mutation in all types of Gaucher patients, as previously reported Korean studies14,15), L444P homozygosity, which was known to may have correlation with chronic neuronopathic type15), was found in 2 non-neuronopathic patients in this study. Further genetic-molecular evaluation for this discrepancy is required.
In particular, because the G46E mutation is a unique form in Korean patients with the non-neuronopathic type14), this mutation may be investigated for further genotype-specific treatment using a chemical charperone. R277C, a novel variant identified in our study, is expected to be a mutation by in silico analysis. In vitro functional analysis can help for the validation. A mutational analysis for the recombination allele will be performed for the patient with only one mutation identified (Patient No. 10).
Although ERT showed limited effects on neurological outcomes, patients with the non-neuronopathic and those with chronic neuronopathic Gaucher disease showed improvements on clinical outcomes following ERT, as demonstrated by our results. Thus, ERT should be recommended for neuronopathic Gaucher disease considering patient's quality of life. However, considering the high proportion of the neuronopathic type in the Korean population, investigations into new therapeutic strategies targeting the nervous system are required.
The disease status of a patient on ERT is defined by subjective symptoms, degree of hepatosplenomegaly, anemia, and thrombocytopenia, biomarkers, and neurological state. However, because existing biomarkers have quantitative limitations to reflect disease activity, and subjective symptoms such as life quality are hard to measure objectively, the development of a new scoring system and biomarkers representing the clinical courses more comprehensively is needed.
In conclusion, Gaucher disease has variable clinical courses, which can be effectively managed with ERT. Considering the high proportion of the neuronopathic type in Korean patients with Gaucher disease, new therapeutic strategies targeting the central nervous system are required. More specific genotype-phenotype correlations should be searched in a greater number of patients, which will help develop genotype-specific treatment modalities.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


