

Introduction
Carnitine is a water-soluble quaternary amine that serves as an essential co-factor in the transport of long-chain fatty acids as acylcarnitine esters across the inner mitochondrial membrane for ╬▓-oxidation1). ╬▓-oxidation of long-chain fatty acids in the mitochondria provides a major energy source in the resting state as well as during fasting and exercise, particularly in heart, liver and skeletal muscle2). This process is particularly important in the early neonatal period, during the transition from a dependence on glucose as the major source of energy in the fetus to the use of fat as the major energy source in the neonate2).
Transient carnitine transport defect usually develops in newborns born to mothers with carnitine uptake defect3). To date, there have been no reported cases of transient carnitine transport defect with prolonged cholestatic jaundice in Korea. Herein, we describe the case of a premature newborn with transient carnitine transport defect associated with prolonged direct hyperbilirubinemia diagnosed by the finding of low free carnitine in the serum and responded dramatically to oral carnitine replacement.
Case report
1. Generals
The patient was a female newborn born by Cesarian section due to placenta previa at 31 weeks of gestation. She was the second child of healthy, non-consanguineous parents; the mother is a 32-year-old teacher and the father is a 35-year-old businessman. There were no specific problems reported during pregnancy, and the patient's Apgar scores were 2 and 7 at 1 minute and 5 minutes, respectively. The birth weight, head circumference and length were 1.620 g, 46 cm and 30 cm, respectively, all within 2 standard deviation. Physical examination upon admission to the neonatal intensive care unit (NICU) was normal, with the exception of minimal chest wall retraction observed at 88% oxygen saturation monitored with pulse oximetry. The patient was placed on nasal continuous positive airway pressure at admission. After one hour, ventilator support was switched to nasal intermittent mandatory ventilation, due to frequent respiratory holding, and the ventilator was turned off at 5 days of age. During her stay in the NICU, the patient showed no symptoms or signs of heart failure or cardiomyopathy, and hypoglycemia (<50 mg/dL in serum) was never observed.
2. Nutrition and growth
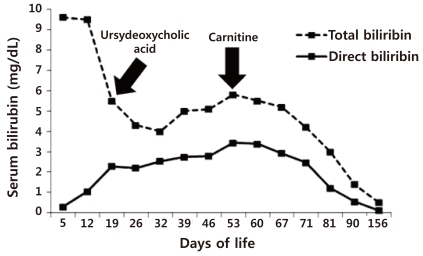
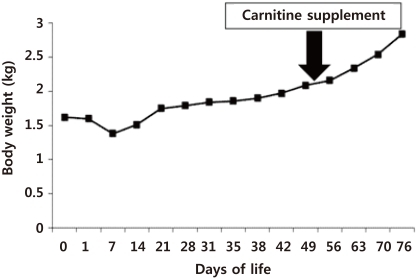
Gavage feeding was started at 48 hours of age, and the first newborn screening test was performed at 72 hours of life. At 12 hours of age, the patient was started on parenteral nutrition, which began to be tapered off at 15 days of life, in consideration of the elevation of direct bilirubin to 1.04 mg/dL (total bilirubin=9.5 mg/dL) from the previous result of 0.29 mg/dL one week earlier. At 19 days of life, lipid infusion at 1.0 g/kg/day was stopped due to the rapid elevation of direct bilirubin to 2.29 mg/dL (total bilirubin=5.5 mg/dL) from the previous result, 1.04 mg/dL (Fig. 1). At that time, she began to receive 12 mL of breast milk with breast milk fortifier and 1.5 mL of medium-chain triglyceride added, making up 70 kcal/kg/day. Although the patient entered full enteral feeding from 26 days of life (24.3 mL per time of breast milk with breast milk fortifier, every three hours, supplying approximately 95 kcal/kg per day), she still showed poor weight gain (Fig. 2). Following carnitine supplementation begun at 53 days of life, the patient gained weight visibly (Fig. 2).
3. Liver function tests
At 19 days of life, the direct/total bilirubin increased remarkably to 2.29/5.5 mg/dL from the previous result of 1.04/9.5 mg/dL one week earlier, and gamma glutamyl transpeptidase (GGT) went up to 189 U/L from the previous result of 160 U/L one week earlier (Fig. 1). Partial parenteral nutrition was then running with lipid and aminoacid being given at 1.0 g/kg/day. Lipid administration was discontinued at 19 days of age, however, due to the elevated direct bilirubin (2.29 mg/dL), ursydeoxycholic acid began to be given (Fig. 1). However, direct bilirubin did not decrease, instead increasing rapidly to 2.35 mg/dL (total bilirubin=5.0 mg/dL) and 2.75 mg/dL (total bilirubin=5.1 mg/dL) at 32 and 39 days of life, respectively, and finally, to 3.44 mg/dL (total bilirubin=5.8 mg/dL) at 53 days of age (Fig. 1). Similarly, GGT and alkaline phosphatase (ALP) increased to 220 U/L and 390 U/L, respectively at 53 days of life from the previous results of 195 U/L and 355 U/L (Fig. 3). However, triglyceride was persistently in the normal range (Fig. 3). After carnitine supplementation was begun at 53 days of life, direct bilirubin, GGT, and ALP began to decrease significantly (Fig. 1, 3). During her stay in the NICU, the patient's serum glutamic oxaloacetic transaminase/prothrombin time and calcium and phosphate remained within normal range (data not shown).
4. Hemoglobin and hematocrit
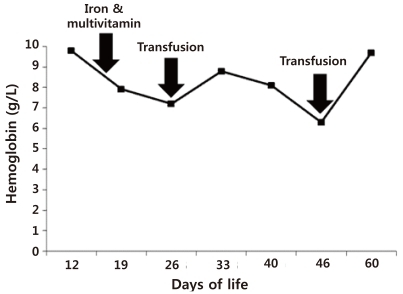
Although iron supplementation (2 mg/kg/day) was started at 16 days of life and was increased to 4 mg/kg/day at 26 days of life, her hemoglobin and hematocrit were only 6.3 g/dL and 18.0% at 46 days of life requiring a transfusion (Fig. 4). At that time, her direct bilirubin and GGT were 2.78 mg/dL (total bilirubin=5.1 mg/dL) and 198 U/L, respectively (Figs. 1, 3).
5. Newborn screening tests
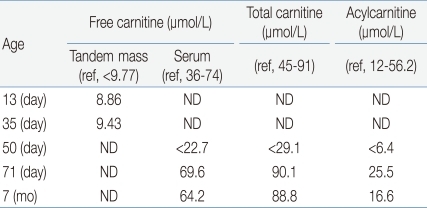
The results of the first newborn screening, performed on the 3rd day of life, showed an abnormality that was suspicious of carnitine transport defect. Further tests conducted at 13 and 35 days of life, using tandem mass test, demonstrated low free carnitine levels, 8.86 ┬Ąmol/L and 9.43 ┬Ąmol/L, respectively (reference range, >9.77 ┬Ąmol/L) (Table 1). To verify carnitine transporter defect, more precise laboratory tests were performed on the patient's serum at 50 days of life. These tests showed low levels of total carnitine, <29.1 ┬Ąmol/L (reference range, 45 to 91 ┬Ąmol/L), free carnitine, <22.7 ┬Ąmol/L (reference range, 36 to 74 ┬Ąmol/L), and acylcarnitine <6.4 ┬Ąmol/L (reference range, 12 to 56.2 ┬Ąmol/L) (Table 1). Carnitine transport defect was thus confirmed, and the patient was started on oral carnitine supplement at 53 days of life (Fig. 1, 2).
6. After carnitine therapy
Oral L-Carnitine supplement of 100 mg/kg per day divided into three feedings was given from 53 days of life. The patient began to achieve sufficient weight gain (Fig. 2), and direct bilirubin began to decrease significantly, reaching 0.82 mg/dL at 81 days of life (Fig. 1). At 71 days of life, carnitine levels including total carnitine, free carnitine and acylcarnitine were reexamined, and had all normalized (Table 1). Considering the possibility of transient carnitine transport defect, carnitine supplementation was stopped at 6 months of postnatal age. Carnitine levels were reexamined four weeks later, and were in the normal range (Table 1). At the present time, the patient is 8 months old. She is growing well and her weight is 8.0 kg (95th percentile).
7. Other tests
The results of the urine organic acid test were non-specific except for a minimal increase in dicarboxylic acid (vanilmandellic acid) and increased ketone bodies (3-hydroxybutyric, acetoacetic acid) were not found in the urine. At 6 month-old, organic acids in the urine were reexamined, and the results showed that the vanilmandellic acid level was normal.
Discussion
The case discussed here is the first confirmed case of transient carnitine transport defect in Korea. Carnitine uptake defect is an autosomal recessive disorder of fatty acid oxidation due to lack of functional carnitine transporter4). Its prevalence has been estimated variably at 1/67,000 to 1/190,000 newborns in Taiwan3,5), 1/40,000 in Japan6), 1/150,000 newborns in Australia7) and 1/250,000 newborns in Germany8), whereas transient carnitine transport defect has been reported in 1/40,000 live births9). However, there has been one case of a cholestatic infant who had transient medium-chain dicarboxylic aciduria with cardiac failure and responded dramatically to carnitine supplementation, with complete resolution of clinical and laboratory abnormalities10). The case presented here is the first reported case of a cholestatic premature newborn with carnitine transport defect confirmed by low carnitine levels in serum and responded to carnitine supplementation.
Carnitine accumulates inside the cells of the heart, skeletal muscle, and kidney4), where it plays an essential role in the transfer of long-chain fatty acids into the mitochondria for ╬▓-oxidation. Hence, even partial deficiency could lead to organ dysfunction11). During periods of fasting, fatty acids are the predominant substrate for energy production4). However, if fatty acid oxidation is defective due to carnitine deficiency, fat cannot be used, and glucose is consumed without regeneration via gluconeogenesis, with hypoglycemia as the possible result4). Moreover, fat accumulates in the heart and skeletal muscle, leading to impaired organ function, in addition to the failure of energy production12). Our patient did not show any metabolic manifestations such as hypoglycemia or hypoketotic hypoglycemic coma, which may be accounted for by the fact that enteral feeding was started at 48 hours of life, after which time, the patient was never in a fasting state, and optimal parenteral nutrition was provided from 12 hours of life. Importantly, infants or young children with Reye's like episodes, including repeated hypoketotic hypoglycemia, should be considered suspicious for carnitine transport defect13).
In carnitine transport defect, cardiac failure is the major presenting manifestation. Over half of the reported cases of this disease first presented between 12 months and 7 years of age with progressive heart failure and generalized muscle weakness14). The hepatic presentation occurs less frequently than the myopathic presentation, because the liver has a separate transporter for carnitine that can usually maintain sufficient levels of carnitine to support ketogenesis15). Our patient had significant and prolonged cholestatic jaundice despite discontinuation of partial parenteral nutrition of lipid and amino acid after two weeks of administration. The explanation for this may be that accumulation of fatty acids in the liver due to carnitine deficiency should adversely affect excretion of bilirubin. Direct bilirubin decreased dramatically following L-carnitine supplementation.
Our patient did not gain sufficient weight after 21 days of life, although she was given more than 85 kcal/kg/day. One likely explanation for her lack of weight gain is that carnitine deficiency may cause some energy depletion. The patient gained weight rapidly once L-carnitine supplementation was started.
Oral L-carnitine (100 mg/kg per day, divided into three separate amounts every day) was given to our patient. She showed a dramatic response, and the effect has been sustained to maintain normal plasma carnitine levels. Oral carnitine supplementation is an effective treatment for transient transport defect, restoring plasma carnitine nearly normal16), and oral carnitine should be given as a regular medication. Additionally, high dose intravenous carnitine may be recommended during life-threatening events, including episodes of metabolic decompensation4,17). Unless cardiomyopathy or metabolic decompensation is present, 100 to 300 mg/kg per day of oral carnitine should be taken regularly throughout the patient's life17).
Carnitine is also known to have a role in red blood cell metabolism, stabilizing the cellular membrane and raising the red blood cell osmotic resistance18). In addition, iron metabolism requires carnitine, a finding supported by previous observations of low serum carnitine concentrations in healthy children with iron deficiency anemia1,3,4). Hence, anemia may be present in carnitine transport defect and the related iron deficiency might worsen the anemia. Our patient was found to have low hemoglobin (6.3 g/dL) and hematochrit (18.0%) at 46 days of age, requiring a transfusion, although she had been receiving iron supplements (2 to 4 mg/kg/day) since 16 days of life. At 46 days of life, she was not yet receiving L-carnitine, and her direct bilirubin had risen to 2.75 ┬Ąmol/L. It is likely, therefore, that the patient's anemia may have been partly related to carnitine transport defect. Since L-carnitine supplementation was started, her hemoglobin level has remained normal.
Transient carnitine transport defect is a rare condition and is generally found in neonates born to mothers with carnitine uptake defect3) or riboflavin deficiency19). Although our patient was not born to a mother with carnitine uptake defect or riboflavin deficiency, and we did not obtain enzyme assays or gene studies with the patient, it is considered highly likely that she had carnitine transport defect, based on confirmation of her low serum carnitine levels, accompanied by a hepatic presentation of remarkable direct hyperbilirubinemia that resolved dramatically with L-carnitine supplementation. And her carnitine transport defect was revealed to be "transient" at 7 months of age by her maintenance of normal levels of serum carnitines four weeks after discontinuation of L-carnitine supplement.
In conclusion, carnitine transport defect is one of the rare treatable metabolic diseases or conditions. Accurate measurement of the levels of free and total carnitine in plasma is necessary in newborns, and oral carnitine supplementation should be introduced in neonates with low plasma levels as soon as samples for metabolic investigations have been collected. Additionally in some NICUs in Western countries, newborn screening tests are performed at 48 hours, as well as 2, 6 and 10 weeks of age. Although the etiological factors for her having transient carnitine transport defect can not be explained exactly, from our experience with this preterm newborn with transient carnitine transport defect, the authors would recommend reexmining newborn screening tests, particularly, in newborns showing unexplained cholestatic jaundice with poor weight gain or profound anemia.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


