


Introduction
Among the chromosomal aberration syndromes, 1q duplications and 18q deletions have been reported separately. 18q deletion is relatively common occurring in approximately 1/40,000 live births1). The phenotype of 18q-syndrome is variable depending on the deleted region and size. Typical forms of 18q-syndrome include developmental delay, microcephaly, facial dysmorphism, growth retardation, congenital aural atresia, hypotonia, and limb anomalies1,2). In patients with distal 18q22.3-23 deletions, dysmyelination and poor differentiation of grey and white matter on brain magnetic resonance image (MRI) have been reported3). Myelin basic protein (MBP) defect, contained in this region is considered responsible for brain dysmyelination. Galanin receptor (GALR1) gene is also located on 18q23, which may be responsible for growth retardation of 18q-syndromes2).
1q partial trisomy is a rare condition and often reported accompanying other imbalances. Clinical features include plagiocephaly, large fontanelles, prominent forehead, down slanting palpebral fissures, low set ears, and others4).
Recent advanced genome wide screening with micro array-based cytogenetic technologies enabled us to detect the genomic imbalances with only a few hundred base pairs. Although interpretations of significance are not always possible, microarray-based comparative genomic hybridization (aCGH) increases the identification of micro duplications or deletions in patients with previously unknown causes of congenital abnormalities and developmental delay5).
We report on a 22-month-old boy presenting developmental delay and other abnormalities who was subsequently diagnosed with a new translocation karyotype of46,XY,der(18)t(1;18)(q32.1; q21.3)[12]/46,XY[152] (7.3% mosaicism) by aCGH, which had not been recognized by routine chromosomal analysis. To our knowledge, this is a newly found partial unbalanced translocation of 1q and 18q.
Case report
The 22-month-old patient was referred to our Pediatric Neurology Department for developmental delay. He was born at 40 weeks of gestation by repeated caesarean with no complications and a birth weight of 2,880 g (10th–25th percentile). He was a second child from a 36-year-old Korean father with mild hearing impairment and 27-year-old Filipino mother in good health. The patient's 4-year-old sister was in normal development. At 9 days old, he was sent to a pediatric cardiologist for heart murmur and diagnosed with a large patent ductus arteriosus (PDA), tricuspid regurgitation (TR) (grade IV) and mitral regurgitation (grade II) by echocardiography. A follow-up echocardiogram at 14 months showed disappearance of the PDA and minimal TR. He had experienced repeated respiratory infection and otitis media, and was once admitted for bronchitis at 14 months old.
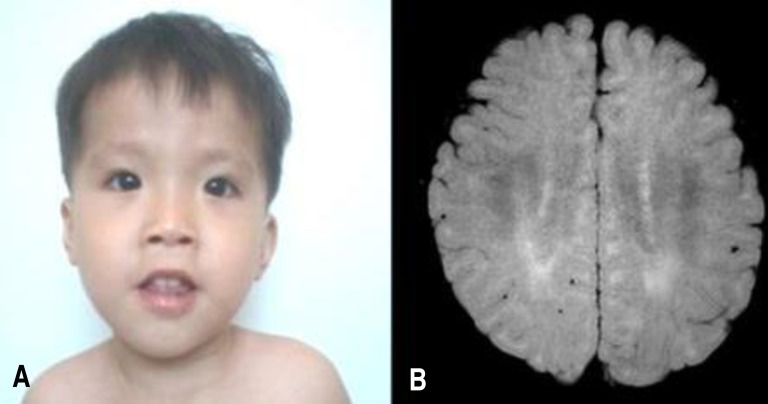
At 22 months old, patient's height was 74 cm (<3 percentile), weight 9.6 kg (<3 percentile) and head circumference 45.1 cm (<3 percentile). On physical examination, he exhibited a mild dysmorphism of the face with frontal bossing, slight narrowing face, down slanting palpebral fissures, low lying ears, mild hypertelorism and a flat nose (Fig. 1A). Both ankles had mild contractures. He was not hypotonic, but tendon reflexes of both knees were slightly increased.
The patient began standing with assistance at 14 months old but could not stand or walk alone. He spoke his first word at 12 months but could only say 3 meaningful words—um-ma (mama), a-bba (papa), a-ppa (painful). He showed generalized developmental delay in standardized Korean infant and child development tests at 22 months old. Total developmental quotient (DQ) was 61.7, and total developmental age (DA) corresponded to 14.3 months old: The DQs (corresponding DAs) of Gross Motor/Fine Motor/Personal Social/Language/Cognitive Adaptive areas were 47.8 (11 months)/60.8 (14 months)/78.2 (18 months)/52.1 (12 months)/69.5 (16 months).
Laboratory findings at 22 months old were white blood cell 11,000/mm3, haemoglobin 12.2 g/dL, and platelet 333/mm3. Blood gases, electrolytes, blood ammonia, lactate/pyruvate, ketone, glucose, thyroid function tests, and serum IgA level were within normal range. Abdominal ultrasonography showed no organomegaly. Skeletal structures were normal. Electro encephalogram was normal. Brain MRI showed mild increased T2 signal in bilateral periventricular white matter (Fig. 1B). Upon otological examination, he was diagnosed with hearing impairment (right, 70 dB; left, 50 dB).
At 39 months old, he had been able to walk unassisted since 29 months; however, he could say only 3–4 meaningful words. His development was assessed as being increasingly more delayed: total DQ 53.7, total DA 18.8 month. Serum insulin-like growth factor-1 was under 25.0 ng/mL (reference, 54–178 ng/mL), and human growth hormone (GH) was 1.3 ng/mL (reference, <0.7–6 ng/mL). At that time, his parents agreed to conduct genetic analysis.
1. Cytogenetic analysis
The patient's first chromosomal analysis was done by GTL-banding with a banding resolution of 550 (20 cells). The second analysis was performed after aCGH analysis and was done by GTG-banding with a banding resolution of 550 (164 cells) (Neodin Medical Institute, Seoul, Korea).
2. Oligonucleotide aCGH
Oligonucleotide-based microarray analysis was performed using a 135K CGX-3 whole-genome microarray (Roche NimbleGen, Madison, WI, USA). Genomic DNA was extracted from peripheral blood, and the patient's DNA and normal control DNA (Human Genomic DNA: Male/Female, Promega, Madison, WI, USA) were labeled with Cy3 and Cy5 with a Roche NimleGen DNA labeling kit. Array hybridization, washing and scanning were performed as specified by the manufacturer (Roche NimbleGen).
3. Fluorescence in situ hybridization analysis
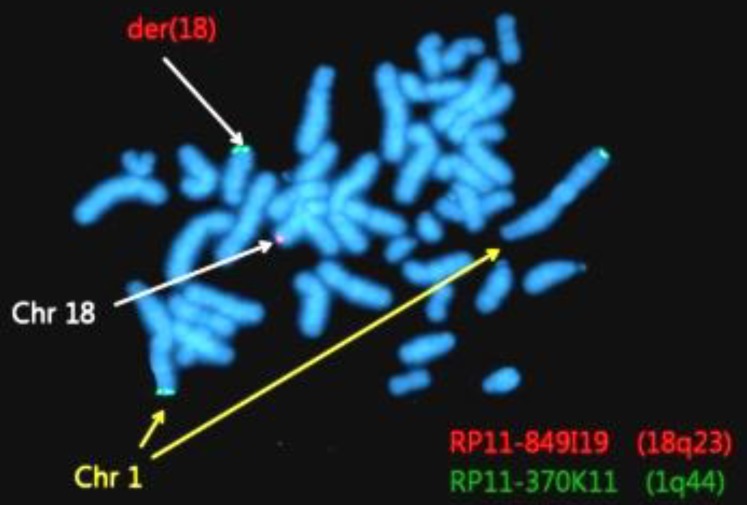
Fluorescence in situ hybridization (FISH) analysis of 150 Metaphase and 50 interphase FISH analysis were done using bacterial artificial chromosome, clone, RP11-370K11(1q44), and RP11-849I 19 (18q23),which located regions of gain or loss.
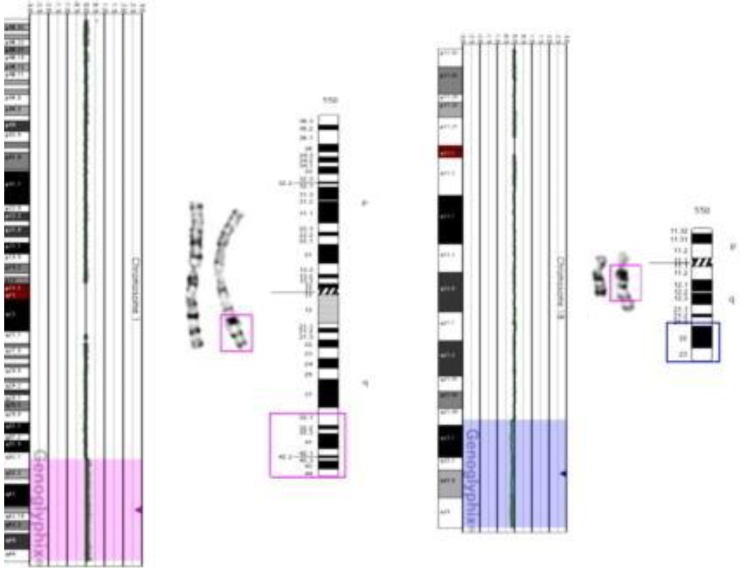
The patient's initial conventional cytogenetic analysis was normal 46,XY. However, abnormal signals in the brain MRI and the patient's dysmorphic face suggested chromosomal aberration syndrome. Subsequent aCGH analysis revealed a subtle copy-number gain of 2,255 oligo probes spanning ~46.38 Mb at 1q32.1 –1q44 (chr1:200,797,519–247,174,728) and a subtle copy-number loss of 874 oligo probes spanning ~46.38 Mb at 18q21.33–18q23 (chr18:58,962,170–76,114,684) (Fig. 2). These findings suggested the possibility of partial duplication of the long arm of chromosome 1 and partial loss of the long arm of chromosome 18.
Repeated chromosomal analysis with a large number of cells revealed unbalanced translocation karyotype of 46,XY,der(18)t(1;18)(q32.1;q21.3)[12]/46,XY[152] (Fig. 3), which indicated the 7.3% of mosaicism.
Abnormalities were confirmed by FISH using 1q44 and 18q23 probes. Among 200 total cells, 185 cells (92.5%) showed 2 red and 2 green signals, and 15 cells showed (7.5%) 3 green signals and 1 red signal (Fig. 4).
As the mother's and father's chromosomes turned out normal (46,XX and 46,XY, respectively), the translocation was a de novo mutation. The patient is currently undergoing rehabilitation therapies and scheduled for GH treatment.
Discussion
Chromosome 18q deletion syndrome is one of the most common deletion syndromes with an estimated frequency of approximately 1/40,000 live births1). This syndrome manifests diverse clinical features depending on deletion size and region. Manifestations include intellectual disability, facial dysmorphism, microcephaly, stenotic ear canals, cardiac, endocrine, genitourinary, immunologic, ophthalmologic, musculoskeletal, and neurologic abnormalities. Neurologic manifestations include developmental delay, hypotonia, seizures, pyramidal and extrapyramidal signs, nystagmus, impaired coordination and white matter abnormalities on brain MRI6).
At the deletion site (18q21.3–23), a total of 34 OMIM genes are located. Two genes of interest at the 18q23 locus are MBP (OMIM 159430, genomic coordinates: 18:74,690,788–74,844,773) and GALR1 (OMIM 600377, genomic coordinates: 18:74,962,007–74,982,097). MBP is abundant in central myelin. In 18q-syndrome, haploinsufficiency of MBP has been considered as related with abnormal T2 signals on brain MRI, suggesting dysmyelination. As galanin is a potent stimulatory factor for GH secretion through growth hormone releasing hormone-dependent mechanisms, defect of GALR1 is responsible for growth retardation in 18q-syndrome7).
The European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations (ECARUCA) database (www.ecaruca.net) lists 2 patients with pure 18q21.3–18q23, without complex rearrangements, among the 22 kinds of all 18q21.3–18q23 deletions. Clinical features include low birth weight, short stature, microcephaly, frontal bossing, hypertelorism, low set ears, small ears, upturned nose, flat nasal bridge, small mandible, hypotonia, single palmar crease, broad hands, club feet, PDA, and tricuspid incompetence. The MRIs were not undertaken. They were diagnosed by GTG-banding.
Duplications of 1q are rare. Complete trisomy 1q has been reported in a few cases and is considered lethal. Partial duplications of 1q are usually caused by unbalanced translocations and present as congenital abnormalities4). Particular breakpoints are 1q21, 1q25, 1q32, and 1q428). Among them, 1q32 to 1qter mostly involves pure partial trisomy 1q. The ECARUCA database lists 11 cases of partial 1q duplications of 1q32–1q44 or 1q32–1qter, with or without complex rearrangements. Until now, three pure inverted or tandem duplications of 1q32–1q44 have been reported4,9). In the region of 1q32.1–1q44, a total of 210 OMIM genes are located. Major features of 1q32-qter are growth retardation, microcephaly, prominent forehead, midfacial hypoplasia, high arched palate, low-set ears, overriding toes, and micrognathia10).
Many of our patient's clinical features were consistent with 18q deletion syndrome but he also displayed some features found in 1q duplication. His cardiac abnormalities, developmental delay, and brain dysmyelination on MRI were corresponded with 18q deletion syndrome. Mild facial dysmorphisms such as frontal bossing, down slanting palpebral fissure, low set ear, flat nose, and growth retardation were corresponded with both 1q duplication and 18q deletion syndromes.
However, relatively mild facial features and attenuated symptoms may owe to his low level mosaicism. His white matter abnormality is a characteristic feature of MBP gene haploinsufficiency at the 18q23 locus. GALR1 gene at the same location may also be associated with his growth retardation.
Our patient was initially suspected of aberration by aCGH analysis, and subsequently confirmed by repeated chromosomal analysis with a large number of cells. As aberration was mosaicism, routine chromosomal analysis could not easily detect the abnormality. This case highlighted again the role of the microarray-based cytogenetic technology to cover insufficiencies of conventional cytogenetic assays.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


