


Introduction
Phelan-McDermid syndrome (PMS) is a rare genetic disorder caused by the deletion of chromosome 22q13.31). Infants or children with this syndrome manifest diverse clinical features, such as seizures, hypotonia, absent or severely delayed speech, global developmental delay, hearing and visual impairment, strabismus, autism-like behavior, cyclic vomiting, normal or advanced growth, and precocious puberty1,2). Patients with PMS also present minor dysmorphic facial features, such as deep-set eyes, poorly formed ears, and a bulbous nose1). The neurological features of this syndrome are nonspecific, and it can be underrecognized along with other genetic diseases that show developmental delays. The mental retardation and developmental delays of these patients are mainly caused by deletions of the SH3 and multiple ankyrin repeat domains 3 (SHANK3) genes1). The SHANK3 gene encodes a structural protein located in the post-synaptic membrane and plays an important role in signal transduction3).
Chromosome analysis is required for evaluation of congenital anomalies and developmental delay. However, it is not suitable for detecting microdeletion. Therefore, fluorescence in situ hybridization (FISH) or multiplex ligation-dependent probe amplification (MLPA) analysis is commonly utilized to identify the microdeletion of the chromosome 22q13.3 region1,2,4). In addition, array comparative genomic hybridization is a useful method for discovering unbalanced translocations5).
Case report
1. Case 1
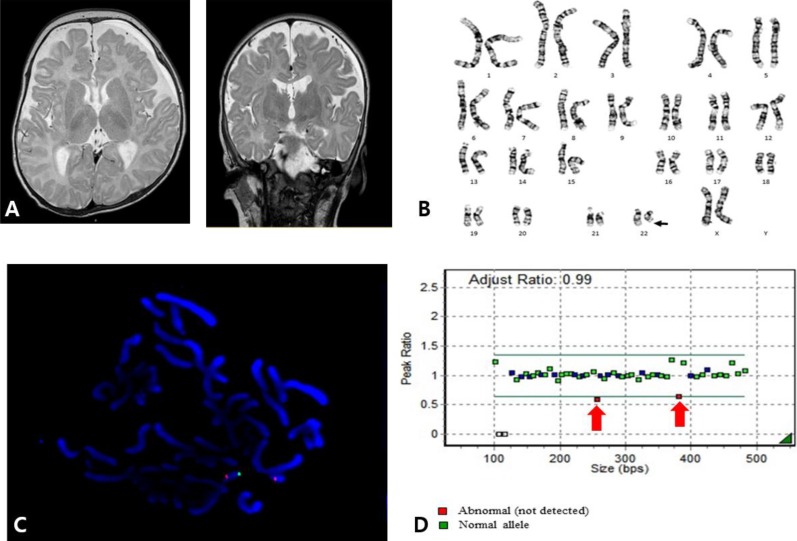
A 4-month-old female, who was born after 39 weeks of gestation with a birth weight of 3,220 g, was referred to our outpatient clinic. An abdominal mass was found during the prenatal ultrasonography, and postnatal ultrasonography determined that it was a multicystic dysplastic kidney although renal function was normal. The patient underwent ligation surgery for patent ductus arteriosus at age 3 weeks. At age 4 months, her eyes could not follow objects, and she displayed hypotonia. Her weight, height, and head circumference were 7.4 kg (50th–75th percentile), 72.4 cm (>97th percentile), and 40 cm (25th–50th percentile), respectively. She had low-set ears, deep-set eyes, wide eyebrows, and a bulbous nose. Brain magnetic resonance imaging (MRI) revealed delayed myelination (Fig. 1A). High amplitude spike discharges from the left frontotemporoparietal area were seen on electroencephalography, but the patient did not present with seizures. Global developmental delay was noted on the Korean infant and child development test performed at age 4 months: her gross motor development level was 1 month, fine motor 3 months, personal-social 2 months, language 2 months, and cognitive-adaptive 3 months.
Chromosome analysis showed 46,XX,del(22)(q13.3) (Fig. 1B). FISH using an HIRA (22q11.2) and ARSA (22q13) probes (Vysis, Abbott Laboratories, Abbott Park, IL, USA) identified the deletion of the ARSA gene (Fig. 1C). In addition, MLPA analysis using the SALSA MLPA P064-C1 kit (MRC Holland, Amsterdam, The Netherlands) revealed the deletion of the SHANK3 gene located on 22q13.3 (Fig. 1D). Neither parent had a chromosome analysis.
At age 8 months, the patient could neither hold her head up nor crawl, but she could roll over by herself and babble. Reaching for and grasping an object was not possible. Global developmental delay was observed in the Denver developmental test: her personal-social development level was 3 months, fine motor-adaptive 3 months, language 6 months, and gross motor 4 months.
Currently, she is 17 months old and still not able to hold her head up. Sitting is possible only when her hands are supported. She is not able to respond to her name. The Bayley developmental test demonstrated a cognitive development level of three months, language 4 months, social 4 months, gross motor 5 months, and fine motor 4 months.
2. Case 2
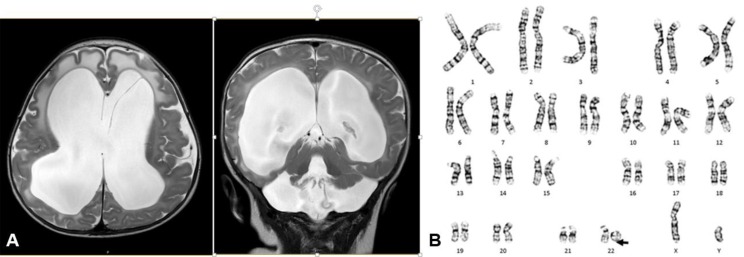
A 4-month-old male was referred to the outpatient clinic for an evaluation of developmental delays. He was born at 38 weeks of gestation with a birth weight of 3,480 g without perinatal problems. There was no family history of congenital anomalies or consanguinity. His prenatal evaluation was unremarkable. Congenital hypothyroidism was diagnosed at age 2 weeks. He was admitted to a local hospital due to recurrent respiratory infections at age 1 month. At 4 months of age, he was not able to control his head or make eye contact with others. His weight, height, and head circumference were 8.7 kg (50th–75th percentile), 67 cm (75th–90th percentile), and 47 cm (>97th percentile), respectively. He showed facial hypotonia, sunset eyes, long eyelashes, dolicocephaly, and relative macrocephaly. Chest computed tomography was performed due to recurrent respiratory infections, and focal narrowing of the mid trachea and left main bronchus between the aorta and pulmonary artery were found. In addition, flexible bronchoscopy showed tracheomalacia and laryngomalacia. There were no cardiac anomalies on echocardiography. Kidney ultrasonography showed right renal pelviectasis. Communicating hydrocephalus with interstitial edema of periventricular white matter was observed on a brain MRI, and this required a ventriculoperitoneal shunt at age 5 months (Fig. 2A). One day after the procedure, heart failure and cardiac arrest occurred, and the patient subsequently died of multiple organ failure. His karyotype was 46,XY,r(22)(p13q13) while both parents had normal karyotypes (Fig. 2B). Deletion of chromosome 22q13.3 was identified by FISH.
Discussion
PMS is a rare disease, and its incidence remains elusive1,2). Facial dysmorphisms, such as long eyelashes, deep-set eyes, ptosis, prominent dysplastic ears, a bulbous nose, and a wide nasal bridge, may indicate this disease2,4). Relatively large fleshy hands, and hypoplastic or dysplastic toenails of the extremities are also present2). Growth velocity is usually normal or accelerated7). Renal abnormalities or cardiac defects are observed in approximately 25% of cases1,2). Gastrointestinal symptoms, such as feeding difficulties, cyclic vomiting, and gastroesophageal reflux, can be associated with PMS1,2). Neurological manifestations include seizures or aggressive behavior1,2).
Although patients with PMS have nonspecific clinical features and diverse clinical courses, it is helpful to perform chromosome analysis to evaluate patients with congenital anomalies and developmental delays. The size of the deleted region in PMS varies widely, ranging from a very small deletion of 100 kb to a very large one of over 9 Mb8). However, the deletion size is not accurately correlated with the severity of the disease. Several genes located in the 22q13.3 region, such as the SHANK3, RAB, RABL2B, and IB2 genes, are thought to be related to neurological features. The SHANK3 gene is located on the distal region to ARSA which is known to cause metachromic leukodystrophy. The SHANK3 protein plays a major role in interlinking between proteins of the postsynaptic membrane and the actin cytoskeleton of the dendritic spine, and it is related to synaptic spine maturation3). Mutations in the SHANK3 gene is also known to be related to neuropsychiatric conditions such as autism spectrum disorder9). Deletions of the ARSA and SHANK3 genes were identified using FISH and MLPA analysis in the present study.
Only one case of PMS has been reported in Korea6). An 18-month-old female with hypotonia and developmental delays was diagnosed by chromosome analysis and FISH using an ARSA probe. The patient's karyotype was 46,XX,del(22)(q13.3), and both parents had normal karyotypes. Brain MRI showed a thin corpus callosum and a decreased white matter volume. This patient underwent rehabilitation therapy, but she was still not able to uphold her head or make eye contact at 3 years (Table 1)6). Global developmental delay, hypotonia, and delayed speech were the main clinical features both found in the previous6) and the present study. Patient 2 who had terminal deletion of 22q13.3 resulting from ring chromosome, showed worse prognosis compared to those who had microdeletion of 22q13.3 region.
Most patients usually have no life-threatening organ dysfunction or events4). However, patient 2 in the present study had severe hydrocephalus urgently necessitating immediate intervention in contrast to patient 1. This patient also had an airway anomaly, which has not been reported in other cases of PMS.
Rehabilitation therapy was applied to increase muscle tone and improve developmental delays. Although progress was slow, obvious enhancement of muscle tone and development was observed.
In summary, this report described the clinical features, outcomes, and molecular genetic data in 2 cases of PMS in Korea. In the event that hypotonia, severe speech delays, mildly dysmorphic facial features, and multiple congenital anomalies involving the kidneys, heart, or toenails are present, PMS should be considered among the possible diagnoses, and molecular cytogentic testing should be undertaken for confirmation.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


