


Introduction
To date, research to increase our understanding of the extensive profiles of microorganisms has been limited. However, the emergence of nonculture sequencing techniques combined with bioinformatics has allowed scientists to better understand the entire microbiome in humans, furthermore, even in the ocean and soil1). The Human Microbiome Project Consortium2) has described the structure, function, and diversity of the microbiome in healthy people in their 2012's report. Humans have remarkably diverse microbiomes in their gut, skin, and vagina. Mammals, including humans, have vast amount of gut surface area, an ideal environment for gut commensals. More than 100 trillion microbes reside in the human gut and continuously interact with the host, resulting in individual immune mutualism, a condition beneficial to both. Microbes exist in homeostasis as commensals, which the host not only tolerates, but also evolves with, to ensure enrichment of beneficial microorganisms3). The primary function of the microbiota is protection, including pathogen displacement, nutrient competition, and production of antimicrobial factors such as bacteriocins or lactic acid. Secondary functions include barrier fortification, induction of IgA, and control of intestinal epithelial cell (IEC) differentiation4).
The microbiome in the human fetus
As the womb has traditionally been considered sterile, an ascending infection from the cervix or vagina could cause adverse events, such as preterm labor5,6). For more than a century, the paradigm was that any bacteria in the womb would be pathologic; therefore, few studies investigated bacteria in a normal, healthy pregnancy. Today, however, high-throughput technology (e.g., 16S ribosomal RNA pyrosequencing or whole-genome-shotgun technique) has made it possible to identify the complete inventory of microbes in the human body. Interestingly, however, the dogma of a germ-free environment within the womb is still maintained. For example, Weng and Walker7), in a recent review, considered the role of the microbiota in immune phenotype programing based on the belief that the acquisition of the microbiota starts when the fetus emerges from the womb. However, recent evidence tells us that the womb is in fact not germ-free8). Because the placenta has been deemed a barrier against pathologic bacteria, it is surprising that the placenta harbors a unique microbiome in healthy, pregnant women9).
Conventional polymerase chain reaction studies in the past decade sporadically identified bacteria in umbilical cord blood during a healthy pregnancy10), as well as in the meconium of normal, healthy infants11). The polymerase chain reaction method also revealed that the placental membrane contained microbes12), including Bifidobacterium and Lactobacillus spp.13). High-throughput analysis has only recently revealed the existence of various dense microbiome communities in the fetus. As neither the amniotic fluid or placenta are sterile, it is assumed that colonization starts during fetal life14,15). Some researchers have presented evidence of placental microbes which may lead to preterm labor and preeclampsia16,17).
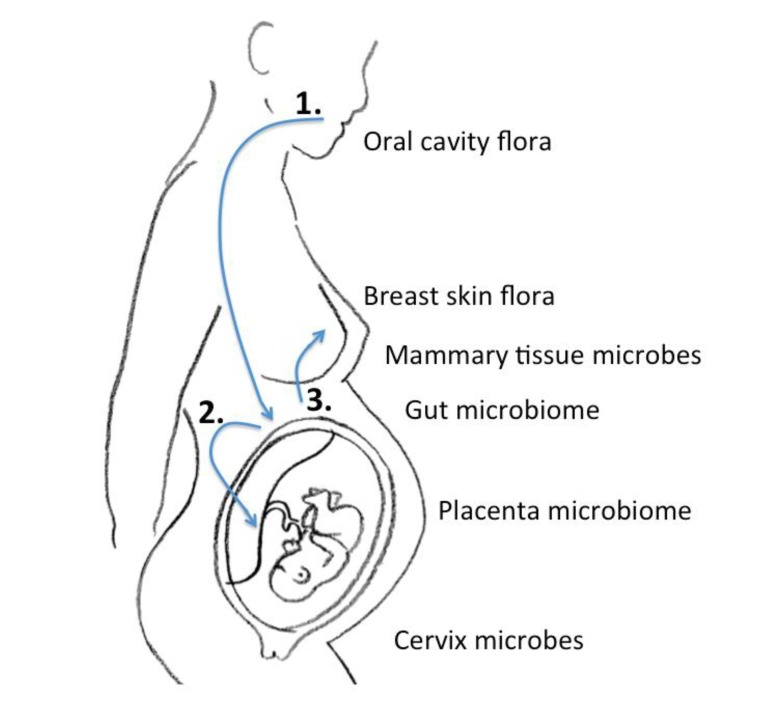
In the era of next-generation sequencing, Aagaard et al.14) analyzed placental commensalism using whole-genome shotgun metagenomics. They found unique placental microbiome commensalism that included bacteria from various phyla, such as Firmicutes, Tenericutes, Proteobacteria, Bacteroidetes, and Fusobacteria. When compared to the maternal microbiomes (oral cavity, gut, cervix, and vagina), the fetal meconium microbiome is more similar to the maternal oral cavity than the vagina. Therefore, it is inferred that bacteria from the mother's oral cavity translocate into the blood stream, then to the placenta, and finally into the fetus (Fig. 1). The traditional ascending pathway from the cervix into the uterus might be limited to pathologic conditions such as chorioamnionitis or early-onset neonatal sepsis. Collado et al.15) found that the amniotic fluid and placental microbiomes were similar in composition. In order to eliminate birth canal contamination, they included cesarean section cases only. Enterobacter, Escherichia, Shigella, and Propionibacterium spp. were dominant. Again, we can infer from these findings that microbes might invade through the IECs, such as dendritic cells, move first to the blood stream, then reach lymphoid tissue, and finally the placenta18). An animal experiment showed that during pregnancy, a 60% increase in intestinal microbes is found in lymph nodes19).
Newborn colonization
Soon after birth, several factors contribute to the initial colonization of the neonatal gut. These include delivery type, breast milk feeding, and antibiotic exposure; caesarean section is associated with decreased microbial diversity and abundance20), breast milk feeding promotes a balanced microbial community and aids in gut development in infants21,22), and antibiotic exposures destroy the normal composition of commensalism23). For preterm infants and full term newborns, breastfeeding or breast milk is strongly recommended for various reasons24). The preventive effect of maternal breast milk against NEC development is specifically true for preterm infants25). Factors in human milk such as immunoglobulins work to protect against hazardous organisms, a passive mechanism from the infant's perspective. In contrast, some factors actively influence the infant immune system, e.g., oligosaccharides as immune modulators26).
Gregory et al.21) recently conducted a study on the role of breast milk ingestion on acquisition of the intestinal microbiome in preterm infants. The preterm infant gut microbiome was greatly influenced by breast milk. Postnatal time, birth weight, gestational age, and nutrition affected the microbial type and diversity. Cong et al.22) stressed similar points, but found that patterns differed between maternal milk-fed infants and those fed human donor milk. Maternal milk conferred optimal balance to the infant microbiome community. The transmission from mother to infant is accomplished by both external and internal transfer. Of course, the flora from the areolar area also contribute numerous microbiota to the newborn. However, the internal breast tissue also harbors microbes that can be transferred. There may be an entero-mammary pathway by which the maternal gut microbiota also transfer to the newborn18).
Microbial role in NEC development
NEC is a major complication in 10% of very low birth weight infants27). The precise mechanism of NEC development is not fully understood. Clinically, diagnosing NEC has definite limitations. Bell's criteria for NEC were first described in the 1970s, when most NEC cases presented in full-term infants28). However, NEC currently targets preterm infants. Furthermore, although the modified Bell's criteria removed Stage I disease in order to eliminate possible overlap with other conditions, the criteria still include sepsis and spontaneous intestinal perforation, as well as NEC itself29). When is the gut most prone to be affected by NEC? What is the most common causative organism? In the early 1990s, Lucas and Cole30) found that exclusively formula-fed infants had 6 to 10 times greater risk of developing NEC. In particular, infants older than 30 weeks of postmenstrual age seldom had the disease. Although this study was published 27 years ago, it is compatible with recent data from the microbiome era. Neu and Pammi31), in a recent review article, briefly discussed unpublished data about NEC incidence. Interestingly, the incidence peaks at 31 weeks of postconceptual age. This is an intriguing finding because it suggests that there must be a certain association between preterm infant gut developmental status and NEC. In other words, it occurs when the gut is ready.
There are numerous recent studies regarding microbial patterns and NEC development. According to one recent meta-analysis32), there is no single causative bacterial agent, but instead a consistent microbiome pattern, that precedes NEC. The pattern is associated with decreased relative abundance of Firmicutes and Bacteroidetes, but increased numbers of Proteobacteria; these findings are associated with strikingly decreased microbial diversity in infants with NEC. These patterns can be considered to reflect “dysbiosis.” This refers to a change from normal, healthy colonization to pathologic conditions. Firmicutes, Bacteroidetes, and Proteobacteria may create an imbalance in intestinal immunity as a result of antibiotic exposure, which results in both an increased risk of NEC and dysbiosis in the gut. The antibiotic exposure group displayed an increased abundance of Klebsiella, Enterobacteriaceae, Proteus, Paenibacillus, Epulopiscium, and Pseudomonas32).
IECs and gut-associated lymphoid tissue constantly interact with microbiota in the lumen. A microbiome-associated molecular pattern (MAMP) stimulates pathogen-recognition receptors that trigger various patterns of immune phenotypes. Dysbiosis dysregulates gut immunity in preterm newborns and eventually results in major morbidities, usually by precipitating proinflammatory reactions. There is abundant evidence for the contribution of the microbiome to a proinflammatory state and NEC development.
(1) A bowel in which Bacteroidetes are decreased secretes a higher level of inflammatory cytokines such as interleukin-633).
(2) The myeloid differentiation primary response gene 88 (MYD-88) is one of 4 immediate adapter molecules that mediate Toll-like receptor (TLR)-MAMP engagement. It is essential for the perception of MAMP and presentation to TLR. When TLR-MYD88 expression is down-regulated, infants are more susceptible to serious infection34). Excessive TLR4 signaling regulates apoptosis, proliferation, and migration of enterocytes, microcirculatory perfusion, and other effects. Normal commensal bacteria exploit TLRs to maintain homeostasis35), but excessive TLR4 expression in dysbiotic intestine can result in NEC36).
(3) Germ-free mice were investigated to determine how the immune system is altered. They showed a decreased level of CD4, CD8, and secretory IgA37,38).
(4) IgA is essential for the maturation of gut immunity. The host gut and commensal microbiota regulate the polymeric immunoglobulin receptor through MAMP. This is regulated by the original colonizing flora. There is evidence that IgA deficiency induces an increase in Proteobacteria39) and that Proteobacteria-specific IgA is responsible for NEC40).
(5) Colonization also influences immune tolerance. Bifidobacterium infantis suppresses the Th2 response to generate oral tolerance in germ-free newborn mice41).
Bacterial diversity and NEC
The intestine is the organ most exposed to macro- and micromolecules and bacteria. Whether lower or higher bacterial diversity exists in NEC is yet to be determined. However, evidence of the former predominates42,43,44). McMurtry et al.45) examined the influence of the microbiome on NEC and found that bacterial diversity was significantly lower in the NEC group. Furthermore, they reviewed subgroup analysis and found a tendency toward a decreasing Shannon index with increasing severity. Microbial diversity in a metagenomic study can be expressed as the Shannon diversity index. This is a quantitative measurement of how many species are present in a specimen. As mentioned above, the fetus shows diverse commensalism in the womb. Disequilibrium affects gut immunity and predisposes to NEC and other diseases.
Microbiome-related epigenetics
Recently, epigenomic studies have been applied to characterize the origins of preterm infant morbidities. Epigenetic mechanisms generate new phenotypic traits. The process takes place during both the prenatal and postnatal period, mainly via DNA or histone modification through enzymatic methylation processes or incorporation of foreign genes. This changes the traits in nuclear architecture so that remodeling of gene expression occurs. Epigenetics are responsible for the development of metabolic syndromes in later adolescent life of intrauterine growth restriction infants through a acquiprocess known as fetal programing46). It mainly involves insulin resistance in these individuals. Additionally, it also involves a gut-microbiota interaction, the “cross-talk.” It deals with preterm birth itself47), infant neurodevelopment48,49), and pulmonary outcomes50). It also confers definite inflammatory traits in the intestine in preterm infants, and can result in high susceptibility to NEC. Remarkably, the prenatal process persists during the postnatal period51). Several studies have shown definitive evidence that colonizing microbiota might induce epigenetic changes48,52). Furthermore, enteral feeding during the Newborn Intensive Care Unit stay greatly influences the gut-microbiota interaction. This epigenome-microbiome cross-talk might be a key player in the development of NEC and other preterm infant morbidities.
Cortese et al.53) demonstrated that probiotic bacteria such as Lactobacillus acidophilus and B. infantis rather than Gammaproteobacteria, such as Klebsiella spp. induced different DNA modifications in a human IEC line experiment. The Klebsiella spp.-treated group showed overrepresented cytoskeleton remodeling. These changes were more sensitive in fetal cell lines than in adult cell lines. They revealed that antenatal corticosteroid resulted in an altered TLR pathway in association with the microbiome. This is quite an impressive finding because the cross-talk takes place very actively in fetal gut to affect individual DNA modification. A typical example of fetal epigenome change is intrauterine growth restriction and insulin resistance. Therefore, future studies investigating host-microbiome interactions in intrauterine growth restriction might be valuable.
Conclusion
The emergence of next-generation pyrosequencing of 16S rRNA has facilitated our ability to analyze microbes in the placenta, cord blood, amniotic fluid, fetal meconium, maternal gastrointestinal tract and mammary gland. The unique and intriguing journey of microbes between a pregnant woman and the fetus is still unclear, however many immunological and epigenetic hypotheses and findings are now being published. Germs continue to cross-talk with the host to form specific individual phenotypes. The cross-talk takes place in the fetus as well as in the preterm infant gut. There is evidence that microbiomes utilize various molecules and DNA to maintain homeostasis; therefore, dysbiosis contributes to the development of NEC in preterm infants.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


