


Introduction
Aniridia means “without iris” in Greek. However, this disorder is panocular, taking its name from the noticeable iris hypoplasia observed in most cases. This anatomical defect ranges from an almost complete absence of the iris with an enlarged and irregular pupil mimicking a coloboma to small slit-like defects in the anterior layer, which can be detected only by transillumination slit lamp biomicroscopy. The effect on vision is similarly variable1).
The aforementioned clinical feature is associated with foveal hypoplasia resulting in reduced visual acuity that is almost always present, as well as with early onset nystagmus. Other associated ocular symptoms include cataracts and late-onset glaucoma2). In addition, such progressive corneal problems are often complicated by the presence of punctate erosion, corneal opacification, and keratopathy2).
Aniridia is caused by mutations in the paired box gene-6 (PAX6), which is essential for eye development3). Aniridia is either isolated without systemic involvement or a part of a syndrome4). Isolated aniridia mainly exhibits autosomal dominant inheritance. The Wilms tumor, aniridia, genital abnormalities, and mental retardation (WAGR) syndrome is a representative syndromic form of aniridia, and is caused by contiguous gene deletion of both PAX6 and the adjacent Wilms tumor 1 (WT1) gene5).
In the current study, we conducted mutational analysis of the PAX6 gene in familial cases of autosomal dominant congenital aniridia and found premature termination of PAX6 protein translation.
Case report
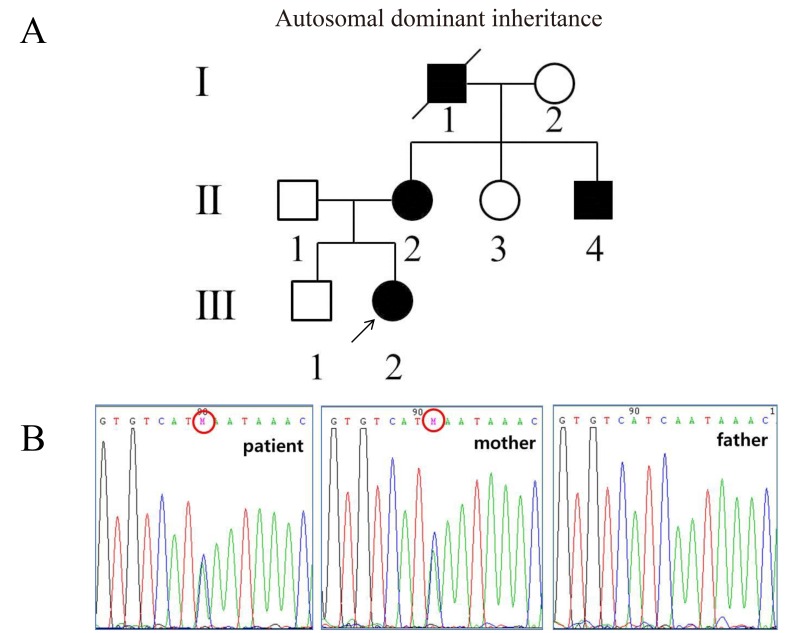
We found a family, in which four members in three consecutive generations were affected by congenital aniridia (Fig. 1A). Peripheral blood genomic DNA was obtained from 6 members of the family (I-2, II-1, II-2, II-3, III-1, and III-2). This study was approved by the Ethics Committee of Jeju National University Hospital and Seoul National University Hospital, and was conducted in accordance with the Declaration of Helsinki. Written consent was obtained for every subject after the nature of the study was explained.
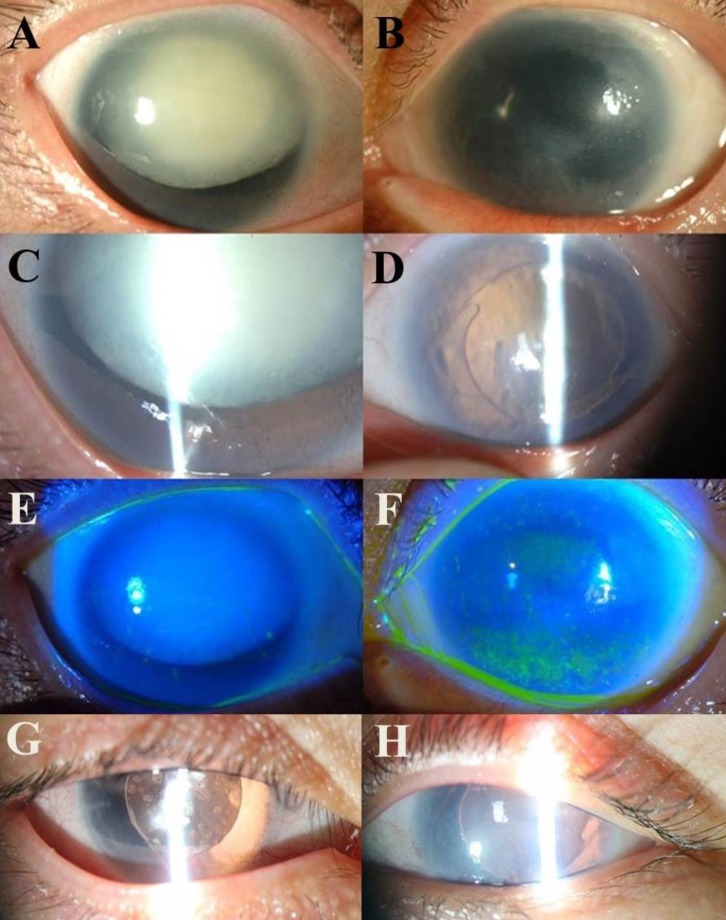
The index case was an 18-month-old girl (subject III-2 in the pedigree). The patient was born via caesarean section at full term gestation and her birth weight was 3.6 kg (50th–75th percentiles). Bilateral aniridia, nystagmus, and foveal hypoplasia were detected at birth. At the age of 18 months, her weight and height were 10.2 kg and 79.7 cm (10th–25th percentiles), respectively. A development screening test using the Korean-Ages and Stages Questionnaires for Infant/Toddler Health Examinations through the National Health Insurance Co. represented the normal developmental milestones. An eye examination revealed complete bilateral absence of irises (Fig. 2). The patient had normal female external genitalia on physical examination.
Subject II-2, currently 34 years old, had bilateral aniridia with nystagmus since birth and secondary glaucoma, lens subluxation, and cataracts that had developed subsequently. She underwent cataract surgeries at the age of 17 and 33 years in both eyes, respectively. She also underwent glaucoma surgery with an Ahmed valve to reduce high intraocular pressure in the right eye at the age of 34 years. Currently, her visual acuity is 0.15 and 0.1 in the right and left eyes, respectively. The findings of the ophthalmologic examination are shown in Fig. 3.
Mutation analysis through direct sequencing of the PAX6 gene revealed a heterozygous nonsense mutation in exon 7 (c.365C>A) causing premature termination at the 122nd codon of PAX6 protein, in all the available affected members (Fig. 1B).
Discussion
It is known that almost all patients with aniridia have mutations in the PAX6 gene. The resultant pathogenic mechanism is haploinsufficiency of PAX6 protein, i.e., loss of function of one allele resulting in a 50% reduction of overall protein activity6). However, a heterozygous mutation in the PAX6 cis-regulatory element (SIMO) that resides in an intron of the adjacent ELP4 gene has been reported in one case7). In our study, a nonsense mutation p.Ser122*, was detected in exon 7 of the PAX6 gene in a family with autosomal dominant congenital aniridia. To our knowledge, the p.Ser122* mutation in exon 7 of the PAX6 gene has not been previously described in Korea, but one case has been reported elsewhere8).
At present, a total of 417 substitution variants in PAX6, mostly with mutations in exons 5, 6, and 9, have been identified. Among the mutations, there are 33.3% nonsense and 17.7% missense mutations, respectively (PAX6 mutation database, http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6). There is no definite correlation between the clinical phenotype and the location of PAX6 mutations9). However, in Korean patients with congenital aniridia, PAX6 mutations have been reported more frequently in exons 7 and 810,11,12). Furthermore, over 90% of the mutations in Korean patients were truncating mutations, including nonsense mutations (50%), splicing errors, deletion and insertion mutations, while missense mutations accounted for 7% of the mutations11).
Glaucoma, which is an important factor for visual acuity in patients with aniridia, generally develops in late childhood, and is rare in infants10,13). Therefore, routine ophthalmologic examination for the detection of glaucoma is important to maintain good vision throughout life. In this report, subject III-2, who was 18 months old, did not have glaucoma and cataract, while subject II-2 had to undergo ocular surgery several times due to cataract and glaucoma.
In addition, in infant patients with aniridia, it may be difficult to differentiate between isolated aniridia and the WAGR syndrome. In young individuals with WAGR syndrome, Wilms tumor and intellectual disability may not be evident and external genitalia are often normal in female patients14). In order to determine who are at high risk for Wilms tumor, genetic diagnosis is essential6).
In conclusion, our familial aniridia cases show differences in the severity of the phenotype according to age. Genetic analysis is important for confirming PAX6 mutations in congenital aniridia and for determining whether an affected member is at a high risk of developing a Wilms tumor. Furthermore, differential diagnosis is needed because aniridia can be associated with a syndromic form in case of a WT1 gene mutation, while EPL4 gene mutations also cause aniridia even though the most common cause for isolated aniridia is a PAX6 gene mutation15). Congenital aniridia is a rare disease in Korea and few cases are reported. Therefore, case collection is important for correlation between the genotype and phenotype of congenital aniridia in Korea. Hence, the authors present this additional case report.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


