


Introduction
The chromosome 11q13 deletion syndrome, the otodental syndrome or oculo-oto-dental syndrome, is a rare but severe autosomal dominant craniofacial anomaly1). So far, only 9 families have been reported to have this syndrome2). The otodental syndrome was first described in 1972 by Levin and Jorgenson. They described that the syndrome is characterized by multiple dental abnormalities that included large, globe-shaped molars, absent or small premolars, delayed eruption of teeth, and high-frequency hearing loss3,4,5,6,7,8,9). This syndrome presented a variable expressivity and penetrance with an autosomal dominant inheritance pattern in familial cases, although some sporadic cases have been also reported2,3,4,5). Common clinical manifestations include grossly enlarged canines and fused molars (globodontia), high-frequency sensorineural hearing loss, and ocular coloboma in some cases2,3).
We report a 1-year-old girl who presented with delayed dentition and other craniofacial dysmorphic features. The array-comparative genomic hybridization (CGH) analysis revealed a deletion in chromosome 11q13.2-q13.3. This report on the otodental syndrome is the first patient in Korea.
Case report
A 1-year-old girl was referred to our neurodevelopmental clinic because of her developmental delay. She was born at 38 weeks of gestational age by vaginal delivery and her weight was 2,090 g (<3rd percentile). The mother was 27 years old at the time of delivery, and the father was 28 years old. The first several months of her life were unremarkable. She could hold her head up at 4 months and roll over at 7 months after birth.
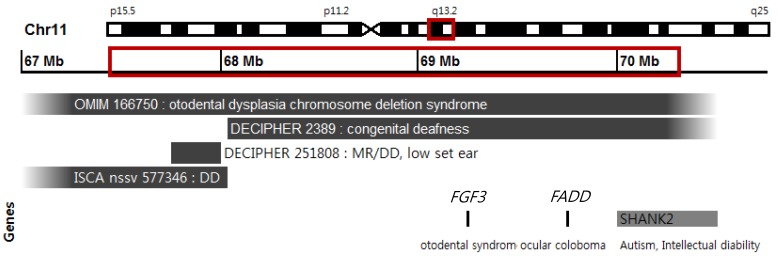
At the age of 12 months, her body weight was 6.6 kg (<5th percentile), her height was 71.5 cm (10thŌĆō25th percentile), and her head circumference was 42 cm (<5th percentile). She had dysmorphic features including ptosis of the left eye, right auricular deformity, microcephaly, high-arched palate, and simian line on the right hand. She presented right microtia (small ear) with shortened upper parts of the auricles. However, the results of the otoscopic examination and auditory brainstem response measurement were normal. During the ophthalmic examination, no visual or structural abnormalities were observed, except the congenital ptosis of the left upper eyelid. Dentition was delayed, but normal teeth were observed upon mandible X-ray examination. She could barely sit with support and could only babble. The Korean Bayley Scales of Infant Development test II had been used to assess her development. According to the test, she had a moderate developmental delay; a Mental Development Index score of 50 (8 months) and a Psychomotor Development Index score of 50 (6 months) at 12 months of age. The results of the metabolic study and thyroid function test were normal. Brain magnetic resonance imaging showed no definite abnormality. The results of the routine chromosomal study were normal (46,XX). However, the array-CGH (NimbelGen 135K CGX-3 Array, Roche NimbleGen, Inc., Madison, WI, USA) analysis revealed a deletion of a 2.75-Mb segment in the region spanning from 67,524,823 to 70,272,728 in chromosome 11q13.2-q13.3 (Fig. 1).
She was reexamined at 17 months of age, when her body weight was 8 kg (<3rd percentile). By this time, she could sit without support, and she could still only babble. The deciduous teeth had not erupted yet. Her left upper eyelid drooped more than it did before, and she underwent ophthalmic surgical treatment.
Discussion
The typical tooth morphology in patients with the otodental syndrome was first described in 1969 by Denes and Csiba from Hungary in a mother and her son2,4,10). The dental abnormalities were classified as those in eruption, number, size, shape, and structure2). Globodontia (large, globe-shaped molars) occurs in both the primary (deciduous) and the permanent dentition. The incisors are not affected, and premolars are often absent. Delayed eruption of the deciduous and permanent teeth is usually observed. Other, less frequent, dental findings include hypoplastic yellow areas in the enamel of the canines and molars, microdontic premolars, numerous supernumerary conoid teeth, deformed maxillary arch with a deep-narrow palate, and odontoma2,4,5,6,7).
In general, sensorineural hearing loss (hearing threshold greater than 25 dB in one or both ears) is more pronounced at frequencies of about 1,000 Hz. The age of onset of hearing loss varies from early childhood to middle age; however, the first symptoms usually occur before the patients are in their 20s. Regardless of the age of onset, hearing loss is bilateral and progressive, but it may plateau by 40 years of age2,4,5,6).
The otodental syndrome is associated with a congenital coloboma of the eye in some cases, and such a syndrome was termed as an oculo-oto-dental syndrome1,2,5,11). Some authors have reported dysmorphic facial features including a long face, full cheek, anteverted nostrils, a long philtrum, and a cleft lip in some patients with this syndrome2,3).
The syndrome was found to show variable expressivity. Dental abnormalities and hearing loss usually coexisted in the patients. However, it was found that some patients developed only sensorineural hearing loss, while others showed dental abnormalities without the hearing loss3,4,5). In our case, we observed a delayed eruption of the teeth; however, sensorineural hearing loss was not detected. Because the onset of hearing loss may vary from early childhood to middle age, we predict that hearing loss may develop later in life.
Our patient had a deletion in chromosome 11q13.2-q13.3 on the array-CGH analysis. The deletion included a 2.75-Mb region spanning from 67.524 to 70.964 Mb (Fig. 1). In 2007, Gregory-Evans et al.1) found that the underlying genetic defect causing the otodental syndrome was the hemizygous microdeletion at chromosome 11q13.3. The smallest deletion, of ~43 kb, was that of the FGF3 gene. This finding suggested that the haploinsufficiency of FGF3 is likely to be the cause of the dental and hearing defects. In addition, the hemizygosity of the FADD gene appears to be responsible for the ocular coloboma formation. The otodental syndrome (OMIM 166750) was named as the chromosome 11q13 deletion syndrome. Recently, recessive mutations in the FGF3 gene have been reported in a new form of severe syndromic deafness, characterized by microtia and microdontia (small teeth) without eye abnormalities (OMIM 610706: LAMM). Some of the affected individuals were reported to have delayed gross motor development12,13). It is possible that the microdeletions may have unmasked a recessive allele of the FGF3 gene on the normal chromosome in individuals with the otodental syndrome1).
Four of the previously described cases (OMIM 166750, DECIPHER 2389, 251808 and ISCA nssv 577346) had overlapping symptoms with our patient. These cases were reported in association with otodental syndrome, ocular coloboma, global developmental delay, and mental retardation (Fig. 1). The Database of Chromosomal Imbalances and Phenotypes using Ensemble Resources (DECIPHER) reported 2 patients, both showing overlapping 11q deletions. Patient 2389 carried a smaller, completely overlapping deletion spanning from 68.072 to 70.307 Mb and had congenital deafness. Patient 251808 had a much smaller deletion between 67.644 and 68.044 Mb and presented clinical features including low-set ears, down-slanting palpebral fissures, speech delay, and mental retardation14).
Wischmeijer et al.14) reported the identification of a cryptic interstitial deletion (3.4 Mb) in chromosome 11q13.2-q13.4 that spans from 67.525 to 70.964 Mb in a child. The child was diagnosed with developmental delay, severe speech delay, moderate/severe mental retardation, and some dysmorphic features. The patient presented a preauricular tag, small low-set ears, ptosis of the upper eyelid, broad nasal bridge, and a short philtrum.
The deleted region in the patient's chromosome in this study included a region of the SHANK2 gene. SHANK2 is a member of a family of genes (comprising of SHANK1, SHANK2, and SHANK3) encoding scaffold proteins. These genes are localized in the postsynaptic density of the excitatory synapses in the brain14,15). Berkel et al.15) identified SHANK2 loss-of function mutations in patients with autism spectrum disorder and mental retardation and suggested a potential role of this gene in the described pathogenesis.
In conclusion, the chromosome 11q13 deletion syndrome (OMIM 166750: Otodental syndrome) shows variable clinical manifestations that involve craniofacial dysmorphology. We recommend performing an array-CGH analysis whenever clinical features of patients may lead to uncertain diagnosis of the otodental syndrome and requiring a functional workup for hearing, ophthalmic examination, and long-term dental care.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


