




Introduction
Abnormalities of potassium channels lead altered characterization of pulmonary arteries in pulmonary arterial hypertension (PAH) [1,2]. Diverse potassium channels, including calciumsensitive (KCa), voltage-gated (Kv) and inward rectifier (Kir) channels, have been involved in pathological changes in PAH [3]. They contribute to vascular tone, proliferation and apoptosis of pulmonary artery smooth muscle cell (PASMC) [4]. Kv channels have been a significant part of membrane potential in pulmonary arteries and are a major mode of action for controlling pulmonary artery tone [4].
Expression and function of Kv channels are regulated by endothelin (ET)-1 and reactive oxygen species (ROS) in PAH. ET-1 decreases Kv channel activity and expression [5]. It has been suggested that ET-1 plays an important role in the development of PAH and stimulates PASMCs proliferation through ROS [6]. ROS, such as hydrogen peroxide, inhibit Kv channel in a rat pulmonary artery. Activation of NADPH oxidase (NOX) and the subsequent production of hydrogen peroxide are involved in the Kv channel inhibition. Impaired ROS pathway is observed in PAH and ROS also increases PASMC contraction and proliferation by increasing intracellular calcium concentration [7-9]. Both ET-1 and ROS cause change of potassium channel expression level in PAH [10,11].
Kv channels are indispensable for the electrical excitability of muscle fibers because they are responsible for cell membrane repolarization after initiation of an action potential [12]. Pharmacological properties of the Kv1.7 channels conduct outward rectifying currents in cardiac tissue [13]. This current plays a central role in cardiac atria repolarization that was largely believed to correspond to the activity of the Kv1.5 channel [13].
However, Kv1.7 channels were poorly understood in the pulmonary artery and Kv1.7 channel was relatively recently identified. We thought that Kv1.7 also have an important role repolarization of PASMCs such as Kv1.5.
The purpose of this study was to investigate whether Kv1.7 channel expressions in the lung tissues of an MCT-induced PAH rats in our model changed and whether this change is caused by ET-1 and ROS pathways.
Materials and methods
This study was divided into 2 parts: (1) an in vivo study and (2) an in vitro study.
1. in vivo study
1) Animal model
Six to ten-week-old male Sprague-Dawley rats (200ŌĆō250 g) were used in this study. The rats were separated into 2 groups: the control (C) group (n=12) and the monocrotaline (M) group (MCT 60 mg/kg, n=12). MCT was introduced by subcutaneous injection. Control rats received an equal volume of phosphate-buffered saline. Study protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Ewha Womans University School of Medicine (IACUC approval No. 14-0266).
2) Hemodynamic measurement
Hemodynamic parameter and right ventricular pressure (RVP) were measured at weeks 2 and 4 after MCT injection. The animals were anesthetized with zoletil (Virbac, Carros, France) and rompun (Bayer Korea, Seoul, Korea) peritoneally. Polyethelene catheters were inserted into RV through the external jugular vein to estimate mean RVP.
3) Organ weight
The right ventricle (RV) was dissected from the left ventricle (LV) and the septum (S) and weighed to evaluate RV hypertrophy. Index of RV hypertrophy was RV/body weight or RV/(LV+S) weight ratio.
4) Pathology
Lung tissue preparation, section, staining and vascular morphometry were performed as previously described [14]. Tissues were fixed with 10 % formalin and embedded in paraffin. Sections were 4 ╬╝m thick and stained with hematoxylin and Victoria blue. Samples were photographed with a digital camera, and the diameter and thickness of pulmonary arterioles were measured more than 20 points in each section by using image-Pro plus 6.0 program. We measured pulmonary arterioles which diameter between 10ŌĆō100 ╬╝m. The medial wall thickness was calculated as (medial wall thickness)=(sum of 2 side of the vessel thickness, ╬╝m)/(outer diameter of the vessel, ╬╝m)├Ś100. These measurements were made directly at ├Ś400 magnification. The number of muscularized pulmonary arterioles was assessed at ├Ś200 magnification and counted in one field of the microscopy.
5) Western blot analysis
Western blot analysis was performed using lung tissues at weeks 2 and 4. Tissues were immediately frozen by liquid nitrogen. The frozen tissues were washed in 4Ōäā phosphate buffered saline and incubated with lysis buffer (Proprep, Intron, Seongnam, Korea) in ice. Quantification of protein was processed with bicinchoninic acid assay, and equivalent proteins were loaded to 8%ŌĆō12% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The separated proteins were transferred to nitrocellulose membrane at 100 V, 2 hours. Antigens were detected using Kv1.7 antibody (Alomone Labs, Jerusalem, Israel), ET-1 antibody (Abcam, Cambridge, UK), endothelin receptor A (ERA) and NOX 4, endothelial nitric oxide synthase (eNOS), glutathione peroxidase (Gpx) 1 antibody and superoxide dismutase (SOD) 2 antibody (SantaCruz Biotechnology, Santacruz, CA, USA). The blots were visualized by enhanced chemiluminescence solution (GE healthcare, Buckinghamshire, UK).
6) Polymerase chain reaction analysis
Total mRNA was extracted and quantified. The mRNA from every sample converted to cDNA (Qiagen, Venlo, The Netherlands). Equal amount of cDNA were used in polymerase chain reaction (PCR). Specific PCR primer pair for the studied genes were; 18s ribosomal RNA, forward 5ŌĆÖ-GTA ACC CGT TGA ACC TT-3ŌĆÖ, reverse 5ŌĆÖ-CCA TCC AAT CGG TAG TAG CG-3ŌĆÖ; Kv1.7, forward 5ŌĆÖ-TCC TTC TAC GGG CTG GGT GC-3ŌĆÖ, reverse 5ŌĆÖ-TTG AGC CGA GCG AGG AAC GA-3ŌĆÖ; Kir6.1, forward 5ŌĆÖ-GAG TGA ACT GTC GCA CCA GA-3ŌĆÖ; reverse 5ŌĆÖ-CGA TCA CCA GAA CTC AGC AA-3ŌĆÖ.
2. in vitro study
1) Cell culture
Primary cultures of PASMCs were isolated from 10-week-old rats as previously described [15]. Briefly, a segment of the main pulmonary artery was excised and placed in DulbeccoŌĆÖs modified Eagle medium containing 10% fetal bovine serum, 1% penicillin and streptomycin. Adventitia was carefully stripped off by a fine forcep. The isolated pulmonary arteries were incubated for 37Ōäā, 20 minutes in 1.5-mg/mL collagenase (Worthington) and papain respectively. After centrifugation, the cells were grown over night at 37Ōäā in a humidified atmosphere with 5% CO2. Few days later, additional complete medium were added.
2) Cell proliferation assay
PASMCs were seeded 5,000 cells/well into 96 well plates and allowed to adhere for at least 18 hours with complete medium. Cells were incubated with serum-free medium containing 1-╬╝M ET-1. After 4 or 24 hours, the number of viable cells were determined using EZ-cytox kit (Daeil Lab Service, Seoul, Korea). Reagent (10 ╬╝L) was added directly to cells in 100-╬╝L medium and after 2-hour incubation at 37Ōäā, absorbance at 450 nm was read using enzymelinked immunosorbent assay (ELISA) reader.
3) ROS detection
PASMCs were seeded 1├Ś104 cells/well in 96 well plate. Intracellular ROS was detected via the dye dihydroethidium (DHE; Molecular Probe, Eugene, OR, USA) or dihydro-dichlorofluorescein diacetate (cm-H2DCFDA); Molecular Probe, Eugene) for superoxide and hydrogen peroxide respectively. After 24 hours of incubation with or without 1-╬╝M ET-1 in incomplete medium, DHE or cm-H2DCFDA were added. The fluorescence intensity of each well was measured with ELISA reader.
Results
1. PAH rat model induced by MCT
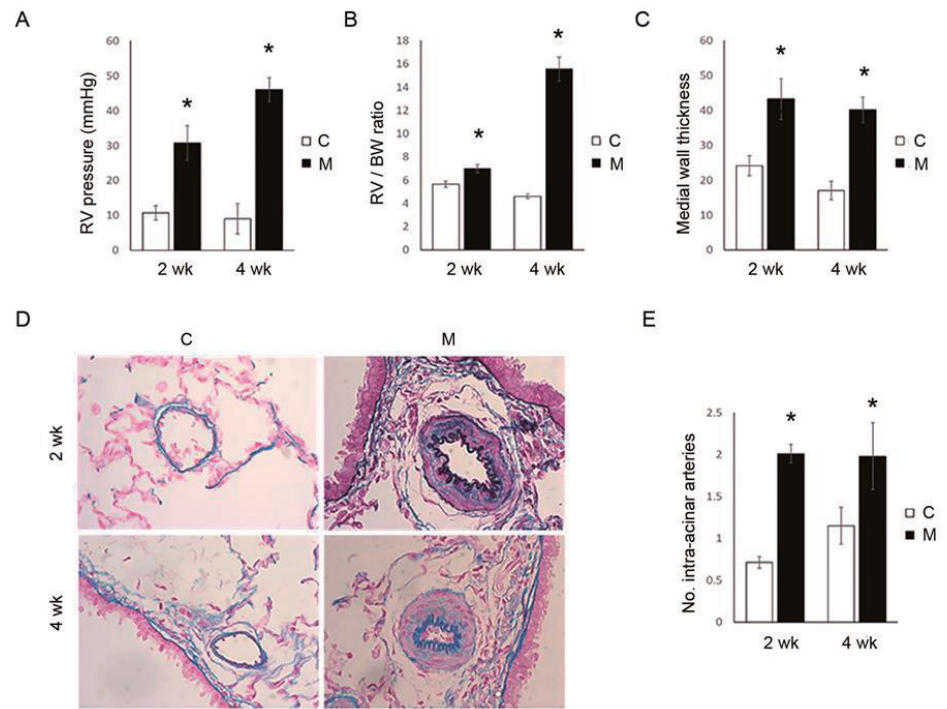
To identify PAH rat model induced by MCT, we investigated mean RVP, RV/LV+S ratio and pulmonary pathology. MCT injection led to PAH with an increase in mean RVP at weeks 2 (C vs. M group; 10.7┬▒2.1 mmHg vs. 30.7┬▒4.9 mmHg, P<0.05) and 4 (C vs. M group; 9.0┬▒4.4 mmHg vs. 46.0┬▒3.5 mmHg, P<0.05) (Fig. 1A). RV hypertrophy as depicted by increment in the ratio of the RV/BW ratio at weeks 2 (C vs. M group; 5.6┬▒0.3 vs. 7.0┬▒0.4, P<0.05) and 4 (C vs. M group; 4.6┬▒0.2 vs. 15.5┬▒1.0, P<0.05) (Fig. 1B). Furthermore, MCT caused pulmonary vascular remodeling as shown by increased medial wall thickness at weeks 2 (C vs. M group; 24.1┬▒2.9 vs. 43.2┬▒5.9, P<0.05) and 4 (C vs. M group; 17.0┬▒2.6 vs. 40.1┬▒3.7, P<0.05) (Fig. 1C, D). The medial wall thickening is concomitant with a reduction of the internal vessel diameter. Increased muscularization in precapillary pulmonary arteries were observed at weeks 2 (C vs. M group; 0.71┬▒0.07 vs. 2.01┬▒0.11, P<0.05) and 4 (C vs. M group; 1.15┬▒0.22 vs. 1.98┬▒0.40, P<0.05) (Fig. 1E). Therefore, it is considered that PAH model was well established in this study.
2. Changes of potassium channel expressions
To investigate which channel expressions are changed in MCT-induced PAH rat model, we conducted microarray analysis (Table 1). Kv9.2, Kv7.2 and KCa1.1 channel expression levels were increased more than 2 times. Kv10.1 (or Kv6.3, depend on alternative splicing) and Kir4.2 channel expression levels were also increased slightly. Kv, KCa and Kir channels showed altered expression.
To confirm that MCT affects Kv1.7 channel expression, we investigated Kv1.7 channel expression level in the lung tissues of MCT-induced PAH rats. Kv 1.7 channel protein expression was decreased in the lung tissues of PAH rats at week 4 (C vs. M, 1.00┬▒0.37 vs. 0.64┬▒0.08, P<0.05) (Fig. 2A). Kv 1.7 channel mRNA expression was decreased in the lung tissues of PAH rats at week 4 (C vs. M, 0.61┬▒0.11 vs. 0.44┬▒0.04, P<0.05) (Fig. 2B).
This is the first report of Kv1.7 channel in MCT-induced PAH rats although Kv1.5 channel has been already well known. We also investigated Kir6.1 channel expression in the lung tissues. The expressions of Kir6.1 channel protein mRNA expression was significantly increased in the lung tissues of MCT-induced PAH rat model (C vs. M, 1.00┬▒0.10 vs. 1.43┬▒0.13, P<0.05) (Fig. 3).
3. ET-1 and NOX4 expression levels in the lung tissues of MCTinduced PAH rat model.
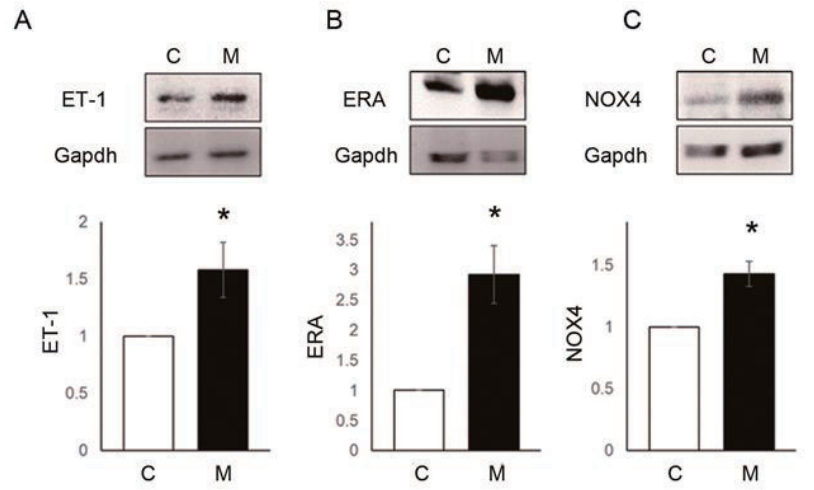
To explain the reasons for the hemodynamic and pathological changes in MCT-induced PAH rat model, we observed protein expression levels of ET-1, ERA and NOX4 in the lung tissues at week 4 (Fig. 4AŌĆōC). Up-regulation of ET-1 (C vs. M, 1.00┬▒0.00 vs. 1.58┬▒0.24, P<0.05), ERA (C vs. M, 1.00┬▒0.00 vs. 2.93┬▒0.48, P<0.05) and NOX4 (C vs. M, 1.00┬▒0.00 vs. 1.43┬▒0.10, P<0.05) were confirmed at the protein expression level in the lung tissues of MCTinduced PAH rat model by western blot analysis. Since NOX4 has been known to induce increased ROS level, we next focused on NOX4 induced by ET-1.
4. NOX4, ROS level and proliferation by ET-1 in PASMCs
To assess whether ET-1 affects PASMCs, we confirmed the changes in ROS enzyme protein expression and ROS level as well as PASMC proliferation in the PASMCs.
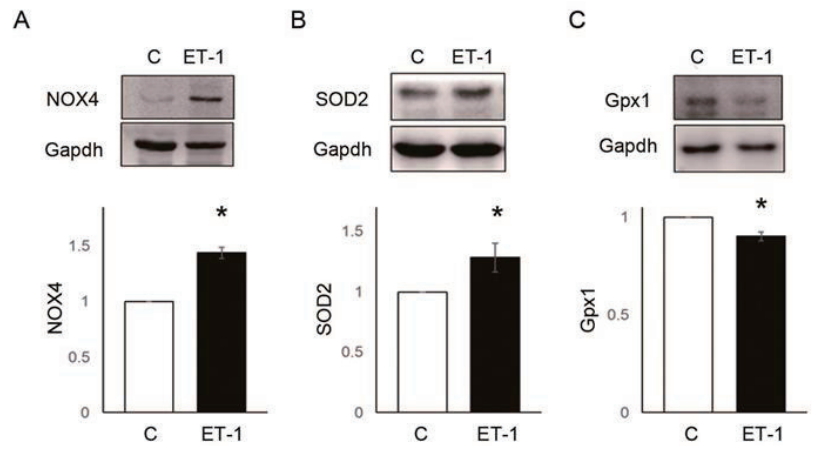
NOX4 (C vs. ET-1 group; 1.00┬▒0.00 vs. 1.43┬▒0.10, P<0.05) and SOD2 (C vs. ET-1 group; 1.00┬▒0.00 vs. 1.00 vs. 1.28┬▒0.12, P<0.05) protein expression level were significantly increased by 24 hours treatment of ET-1 in PASMCs (Fig. 5A, B). However, Gpx1 was decreased by ET-1 (C vs. ET-1 group; 1.00┬▒0.00 vs. 0.90┬▒0.02, P<0.05) (Fig. 5C). Also, ROS levels were significantly increased by ET-1. Hydrogen peroxide (C vs. ET-1 group; 1.00┬▒0.00 vs. 1.48┬▒0.16, P<0.05) (Fig. 6A) and superoxide (C vs. ET-1 group; 1.00┬▒0.00 vs. 1.20┬▒0.12, P<0.05) (Fig. 6B) were increased by ET-1 in PASMCs. As a response to oxidative stress, we expected up-regulation of PASMC proliferation. Proliferation was significantly increased by ET-1 in 24 hours after treatment. Based on these data, it is possible that treatment of ET-1 increased PASMC proliferation by increasing NOX4 and ROS level.
Discussion
Our novel finding is that potassium channels, Kv1.7 and Kir 6.1 channels, are involved in PAH. Elevated ET-1 and Kv1.5 channel levels in the PAH were well known and have been confirmed by a number of studies [16]. However, other potassium channel expression levels in the PAH and ROS enzymes in the PASMCs remain less well defined. The main purpose of this study was to investigate the possibility of the involvement of potassium channels in the pathophysiological changes through ROS and ET-1 in MCT-induced PAH rats.
The results are as follows. First, MCT caused PAH. There was an increase in RVP, pulmonary arteriole medial wall thickness and the number of intra-acinar arteries. Second, potassium channel expression levels were changed in the lung tissues of MCT-induced PAH rats. Altered potassium channels were detected including Kv9.2, Kv7.2, KCa1.1, Kv10.1. Kv6.3 and Kir4.2 by microarray analysis. Decreased Kv1.7 channel mRNA and protein expressions levels were noticed. Increased Kir 6.1 channel mRNA expression level was also observed. Third, ET-1 significantly increased NOX4, SOD2, and ROS protein levels except Gpx1 in the PASMCs.
Therefore, we made the following conclusions: (1) There is a possibility that altered Kv1.7 and Kir6.1 channel expression levels were caused by ET-1 and NOX4 in the lung tissues of MCT-induced PAH rats in our model and (2) ET-1 caused oxidative stress via NOX4, SOD2, and Gpx1 in the PASMCs. Therefore, not only do ET-1 and ROS but also potassium channels probably play a critical role in the pathogenesis of MCT-induced PAH in rat models.
We have attempted to identify the possible mechanisms of altered potassium channel expressions in PAH. We think that potassium channel expression levels are changed by ET-1 and ROS. Firstly, we confirmed that ET-1, ERA and NOX4 protein expression levels were increased in the lung tissues of MCT-induced PAH rats. ET-1 is considered one of the causes of PAH by ROS disruption. In 2001, Wedgwood et al. [14] reported that ET-1 increased ROS in PASMCs. This is consistent with our results. In 2001, Shimoda et al. [17] published that ET-1 (10-8 M) caused significant inhibition of Kv current in human pulmonary arterial myocyte as well as pulmonary artery constriction by ET-1 due to myocyte depolarization by way of the inhibition of Kv. In 2008, in a study by Whitman et al. ET-1 mediates hypoxia-induced inhibition of Kv channel expression in the pulmonary arterial myocytes [5]. In a 2012 study by Wong et al. [18], found that ligands including ET-1 activate the generation of ROS and it leads to the oxidation of target molecules and progresses the disease.
Next, we tried to identify which enzymes were involved in the ROS generation by ET-1 in PASMCs. We identified that superoxide and hydrogen peroxide levels were increased by ET-1 in PASMCs and NOX4, generating superoxide, was also increased by ET-1 in PASMCs. However, Gpx-1, scavenger of hydrogen peroxide, protein expression level was decreased in PASMCs. There have been several studies that show that ROS modulates Kv channel activity and expression level. In 2006, in the study of Cogolludo et al. [10], membrane permeable hydrogen peroxide inhibited Kv channel currents and induced a contractile response. Also, activation of NOX and the subsequent production of hydrogen peroxide are involved in the Kv channel inhibition in rat pulmonary arteries [10]. Furthermore, an imbalance between the oxidant and the antioxidant is critical for the pathogenesis of PAH as the lung is directly exposed to oxygen unlike the other organs [19]. In 2006, Waypa et al. [20] also reported that ROS may increase hypoxia. However, there was a report to describe opposite role of ROS. In 1995, Weir et al. [21] found that antioxidants inhibit whole K+ channel current and oxidants enhance them. In our current study, we observed increased ROS and increased expressions levels of NOX4 and SOD2 in the lung tissues of PAH rats and ET-1 treated PASMCs. Increased expressions of NOX4 had been previously reported in the lungs of PAH rats [22]. It has also been reported that PASMCs are also enriched in SOD2 and that hydrogen peroxide inhibits Kv channels in the arteries [23]. Therefore, we thought that increased ROS might mediate the Kv channel downregulation and other potassium channels. Increased oxidative stress in the lung tissues and PASMCs play a crucial role in the pathogenesis of MCT-induced PAH.
Dysfunction of ion channels is a major reason for increased pulmonary arterial pressure in PAH patients [24]. Several reports have emphasized the importance of potassium channels in the PAH [25-27]. Both in animal models as well as patients with PAH display abnormal potassium channel expressions when compared to non-PAH PASMCs [28]. Our previous KCa3.1 study in an MCT-induced PAH model had the following results: KCa3.1 channel expression level was increased in the lung tissues and pulmonary arterioles in the medial wall. KCa3.1 channel expression level depends on intracellular cyclic AMP level in PASMCs [14]. Potassium channels have a role in maintaining vascular tone by moving potassium ion, and according to their electrochemical gradient, by keeping resting membrane potential in vascular smooth muscle cells. As a result of potassium channel downregulation, potassium efflux is attenuated and membrane depolarization is sustained. The precise clinical effects of Kv1.7 channel is not clear on PAH although Kv1.7 channel mRNA expression level is high in human PASMCs [29]. Decreased Kv channel expression or function results in membrane depolarization, increased intracellular calcium concentration and vasoconstriction in PASMCs. Down regulated Kv1.5 channel exhibits in PASMCs from idiopathic pulmonary hypertension patients when comparing with PASMC from non-idiopathic PAH patients [30,31]. It causes vasoconstriction and proliferation of PASMCs which is a major contributor to the development of pulmonary vascular remodeling [29].
Kir channels also involved in setting the resting membrane potential [32]. Kir channels play a role in the regulation of endothelial proliferation by fibroblast growth factor and it increases endothelial Kir current in human umbilical vein endothelial cells. In our experiment, Kir6.1 channel expression level was increased by ET-1 in PASMCs. Increased Kir 6.1 channel expression was an unexpected result because Kir channel activation raise extracellular potassium ion concentrations and induce vasodilation [33]. It is very hard to explain why Kir 6.1 channel was increased but it can be a compensatory mechanism. Some pathological changes can be reversed at some point during the progression of the disease. Excessive vasoconstriction occurred in our MCT model so the body system efforts to induce vasodilation and increase Kir6.1 channel as a part of a means to overcome the disease.
The limitation of this study is that we did not confirm all potassium channel expression levels in our animal model and PASMCs because there were too diverse kind of potassium channels. To our knowledge, this is the first report of significance of Kv1.7 channel expression changes in the lung tissues of PAH. Furthermore, our findings demonstrate the importance of potassium channels in PAH. Considering the microarray results, many potassium channels are crucial in developing PAH. Selective inhibitors for Kv channel and oxidant signaling molecules will be helpful in the development of therapeutics. Also, precise mechanisms among ET-1, ROS and Kv1.7 would be useful for designing preclinical trials in the use of antioxidants in the treatment of PAH.
In conclusion, potassium channels, ET-1 and ROS have a crucial role in MCT-induced PAH rat models. We carefully suggest that ET-1 cause abnormalities in potassium channel and ROS, and these changes are important in the pathogenesis of PAH.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


