

Introduction
Progressive high vascular resistance becomes pulmonary arterial hypertension (PAH) leading fatal conditions and there is not any certain treatment. The possible mechanisms of PAH are an enhancement of the proliferation and suppression of the apoptosis of pulmonary arterial smooth muscle cells1,2). The role of periarterial proinflammatory cytokines has recently been implicated3,4) as the pathogenesis of pulmonary hypertension. Resistance to apoptosis has been shown to induce vascular proliferation in experimental cell culture5). Aberration in the normal balance between the proliferation and the apoptosis of smooth muscle contributes to vascular remodeling in PAH6).
Tumor necrosis factor (TNF)-α can exacerbate inflammatory reactions that occur in numerous disease conditions. A central role of TNF-α is to induce other proinflammatory mediators such as interleukin-6, granulocyte-macrophage colony-stimulating factor and also other chemoattractive peptides. Infliximab is a chimeric (mouse/human) immunoglobulin G1 monoclonal antibody that binds to the proinflammatory cytokines, TNF-α, with high affinity and specificity. Infliximab improves endothelial dysfunction in heart failure patients7) and improves PAH in sarcoidosis patients8).
There are studies that examine the changes of pulmonary vascular tissue and gene expressions of inflammatory cytokines after administration of TNF-α antagonist in PAH rat models9). The monocrotaline-induced PAH rat model mimics PAH in humans and is applicable in the studies about the effects of several therapeutic agents such as anti-inflammatory and antiproliferative agents10-13). The purposes of this study were to investigate the changes of architecture in the pulmonary arteries and right ventricular pressure (RVP) and to explore the effect of infliximab in monocrotaline induced PAH. We chose six cytokines that involved vascular inflammation, tissue remodeling and vascular constrictions, and dilations. The expressions of six genes (TNF-α, endothelin-1 [ET-1], endothelin receptor A [ERA], endothelial nitric oxide synthase [eNOS], matrix metalloproteinase [MMP] 2, and tissue inhibitor of matrix metalloproteinase [TIMP]) were studied in a monocrotaline induced PAH rat model by reverse transcription-polymerase chain reaction (RT-PCR).
Materials and methods
1. Materials
Six-week-old male Sprague-Dawley rats, weighing between 200 to 250 g, were used for this study. All rats were housed in climate-controlled conditions with a 12 hours light: 12 hours dark cycle and had free access to chow and water.
2. Methods
Pulmonary hypertension was induced by subcutaneous injection of 60 mg/kg monocrotaline (Sigma Chemicals, St. Louis, MO, USA) dissolved in 0.5 N HCl solution. The rats were grouped as follows: C group (n=27), subcutaneous injection of normal saline (0.1 mL/kg); M group (n=36), subcutaneous injection of monocrotaline; M+I group (n=27), subcutaneous injection of monocrotaline (60 mg/kg) plus single subcutaneous injection of infliximab (5 mg/kg).
The rats were sacrificed after 1, 5, 7, 14 and 28 days. Lung tissues were removed and immediately frozen at -70℃ for enzyme analysis, postfixed in 10% formalin and processed routinely for paraffin embedding.
The animal studies were performed after receiving approval of the Institutional Animal Care and Use Committee (IACUC) at Ewha Womans University (IACUC approval No. 10-0141).
3. Ventricular weight
The rats were weighed and observed for general appearance during the study period. The animals were sacrificed after 1, 5, 7, 14 and 28 days after injection and the hearts and lungs were rapidly removed. The weights of the right ventricle (RV), left ventricle and interventricular septum (LV+S) were measured. The ratio of organ weight to body weight was calculated. The RV to LV+S ratio [RV/(LV+S)] was used as an index of right ventricular hypertrophy (RVH)14).
4. Histologic findings of pulmonary arteries
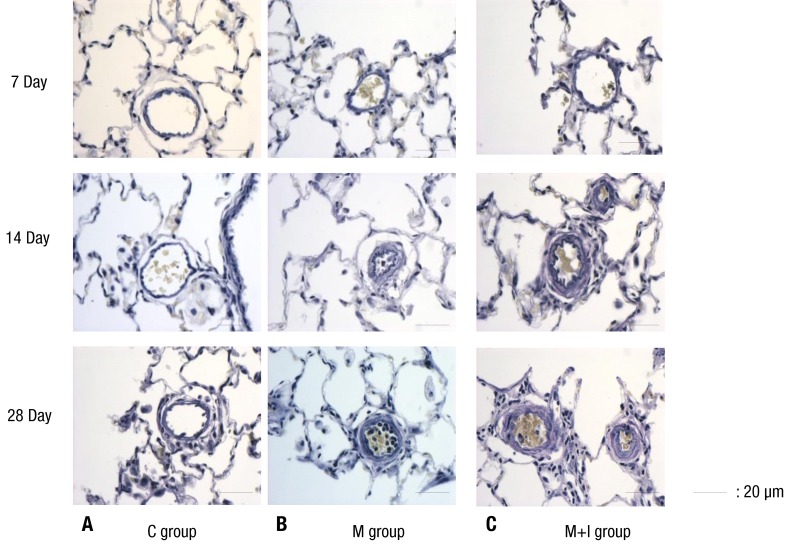
Lung tissue was fixed with 10% buffered formalin and then embedded in paraffin. Sections were performed by 3 µm-thick hematoxylin-eosin and elastic van-Gieson stains to evaluate histopathologic changes of pulmonary blood vessels. More than 20 images of pulmonary arterioles (25 to 100 µm diameters) per tissue section at a magnification of ×400 were captured using a microscopic digital camera and analyzed using an image analysis program (analySIS, Olympus Soft Imaging Solutions, Singapore, Singapore). The external diameter (D) and medial thickness on either side (M1 and M2) were measured along the shortest diameter. The medial wall thickness was expressed as follows: % wall thickness=[(M1+M2)/2/D]×100. In addition, the number of intra-acinar muscular pulmonary arterioles was counted14).
5. Hemodynamic parameter
The animals were placed in the supine position and instrumented with an arterial pressure line (Physiological Pressure Transducer, MLT1199; AD Instruments, Oxfordshire, UK) when sacrificed. Hemodynamic parameters were recorded on day 1, 5, 7, 14 and 28. The catheter was placed in the RV to estimate mean RVP.
6. Microarray analysis of lung tissues for the selection of 6 gene expressions
Total RNA was extracted from rat heart and lung tissues using TRI reagent (MRC, Cincinnati, OH, USA) according to the manufacturer's instructions. Each total RNA sample (10 µg) was labeled with cyanine (Cy5) conjugated dCTP (Amersharm, Piscataway, NJ, USA) by a reverse transcription reaction using reverse transcriptase, SuperScrip ll (Invitrogen, Carlsbad, CA, USA). The labeled cDNA was then concentrated using an ethanol precipitation method. The concentrated Cy5 labeled cDNAs were resuspended in 10 µL of hybridization solution. Afterward the labeled cDNAs were placed on Roche NimbleGen Rat genome 12-plex array (Roche NimbleGen, Madison, WI, USA) and covered by a NimbleGen H12 mixer (Roche NimbleGen). The slides were hybridized for 12 hours at 42℃ by the MAUI system (Biomicro systems, Salt Lake, UT, USA). The hybridized slides were washed in 2× saline sodium citrate, 0.1% SDS for 2 minutes, 1× saline sodium citrate for 3 min, and then 0.2× saline sodium citrate for 2 minutes at room temperature. The slides were centrifuged at 3,000 RPM for 20 seconds to dry.
The normalized and log transformed intensity values were then analyzed using GeneSpring GX 7.3.1 (Agilent Technologies Inc., Santa Clara, CA, USA). Fold change filters required that the genes be present in at least 150% of controls for up-regulated genes and lower than 66.67% of controls for down-regulated genes.
7. mRNA detection by RT-PCR
1) RNA extraction
The rat lung tissues were kept at -70℃. We extracted total RNA from 50 mg of rat tissue using Trizol reagent (Invitrogen). The process was as follows: After adding 1 mL of Trizol reagent in the tissues of the rat, the tissue was crushed using homogenizer and then centrifuged at 14,000 RPM and 200 µL of chloroform was added. After mixing the supernatant fluid with the same amount of isopropanol, the reaction was done at room temperature. After a reaction, we centrifuged total RNA at 14,000 RPM; precipitate was obtained and washed by 75% ethanol. The total RNA was examined by Bioanalyzer 2100 (Agilent Technologies Inc.) and stored at -70℃.
2) cDNA synthesis
One µg of the RNA was synthesized using AB High Capacity RNA-TO-cDNA Kit (Applied Biosystems, Foster City, CA, USA). At first, 1 µg of RNA volume was mixed with dextrose water to total volume 9 mL, after then spinned down in ice for 1 minute at the temperature of 37℃ for 60 minutes. Finally, it was heated at 95℃ for 5 minutes. Synthesized cDNA was kept at -20℃.
3) RT-PCR analysis
RT-PCR was performed in triplicate in 384-well plates. The resulting first-stranded cDNA was normalized by the glyceraldehyde 3-phosphate dehydrogenase gene. The normalized cDNA was used for the PCR procedure as a template.
The specific primers for rat TNF-α, ET-1, ERA, eNOS, MMP2 and TIMP were shown in detail in Table 1.
The PCR reaction was conducted in a final volume of 20 µL containing cDNA 0.5 µL, 0.8µL of primer (10 pM/µL) and 2× SYBR Green PCR Master Mix (Applied Biosystems), which included the HotStarTaqt DNA-polymerase in an optimal buffer 0.4 µL (5 U/µL), the dNTP mix (with dUTP additive) 1 µL (each 2.5 mM). All primers were amplified using the same conditions. Thermal cycling conditions 50℃ for 2 minutes and 95℃ for 10 minutes followed by 40 cycles of 95℃ for 30 seconds and 60℃ for 30 seconds and 72℃ for 30 seconds. In order to exclude the presence of unspecific products, a melting curve analysis of products was performed routinely after finishing amplification by high resolution data collection during incremental temperature increase from 60℃ to 95℃ with a ramp rate of 0.21℃/sec. RT-PCR cycle numbers were converted to gene amounts (ng) on the basis of the equation. The RT-PCR analysis was performed on an Applied Biosystems Prism 7900 Sequence Detection System (PE Applied Biosystems). The amount of each gene expression was revised by relative quantification assay14).
Results
1. Heart weight
There was a significant increase in RV weight on days 14 and 28 in the M group compared with the C group. In M+I group also showed the increase of RV weight on day 1, 7, 14, 28.
RV/LV+S was significantly increased on days 7, 14 and 28 in the M group compared with the C group. However, there were no significant differences between the M and the M+I group.
The weight of LV+S was significantly lower in the M group and the M+I group compared with the C group on days 5, 7 and 28 (Table 2).
2. Histologic findings
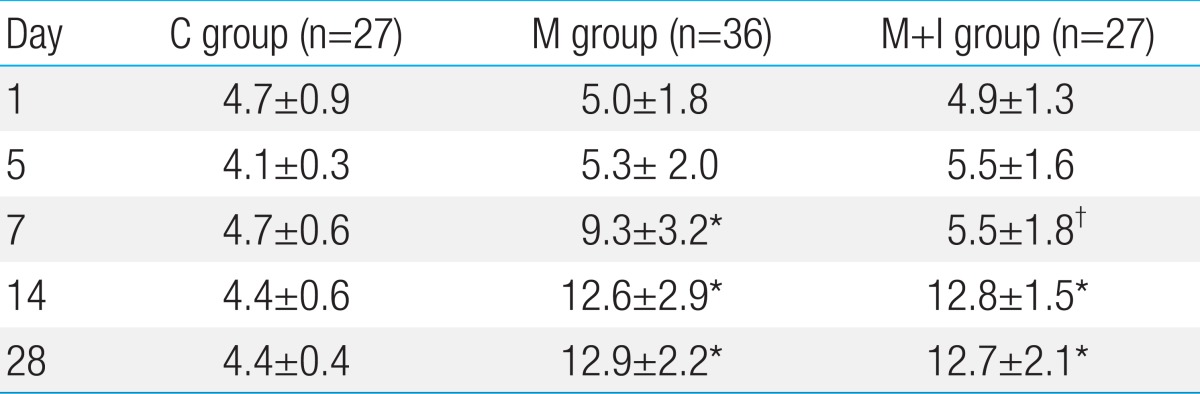
The medial wall thickness in the M+I group was significantly attenuated on day 7 compared with the M group (5.5%±1.8% [M+I] vs. 9.3%±3.2% [M], P<0.05) (Table 3, Fig. 1). The medial wall thickening in the pulmonary arterioles increased in the M and M+I group compared than the C group on days 7, 14 and 28.
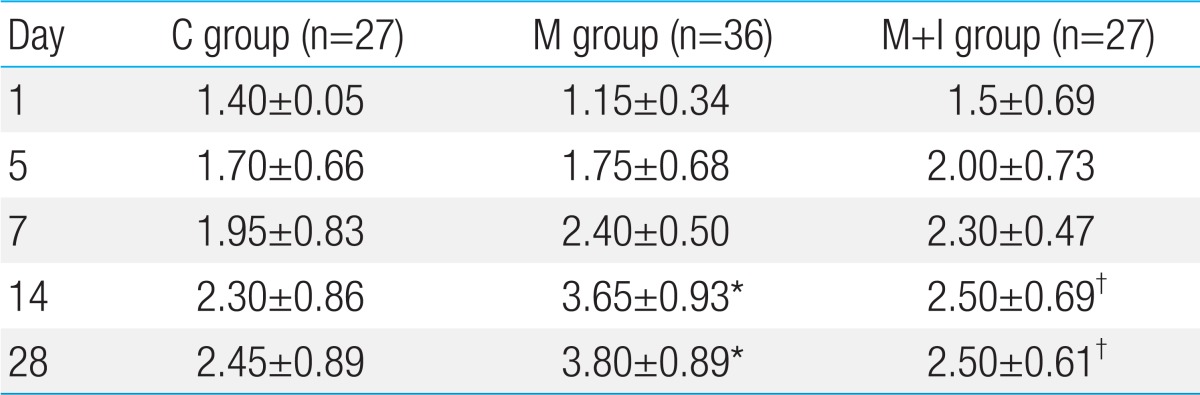
The number of intra-acinar muscular arteries was higher in the M group compared with the C group. The number of intra-acinar muscular arteries was significantly decreased in the M+I group on days 14 and 28 (Table 4).
3. Mean RVP
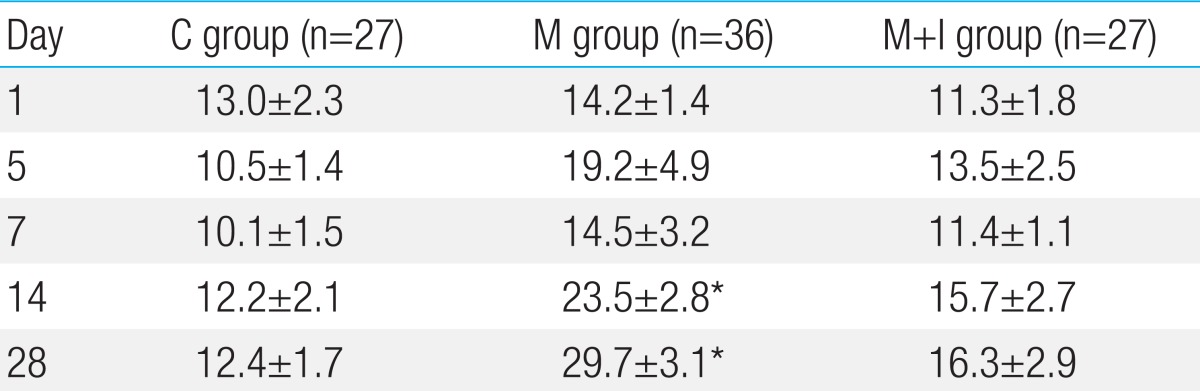
The mean RVP was significantly higher in the M group than the C group on days 14 and 28. The mean RVP showed no difference between the C group and the M+I group. The mean RVP was decreased in the M+I group compared with the M group on days 14 and 28, but it was not statistically significant (Table 5).
4. Gene expressions of TNF-α, ET-1, ERA, eNOS, MMP2 and TIMP
1) Gene expressions of TNF-α
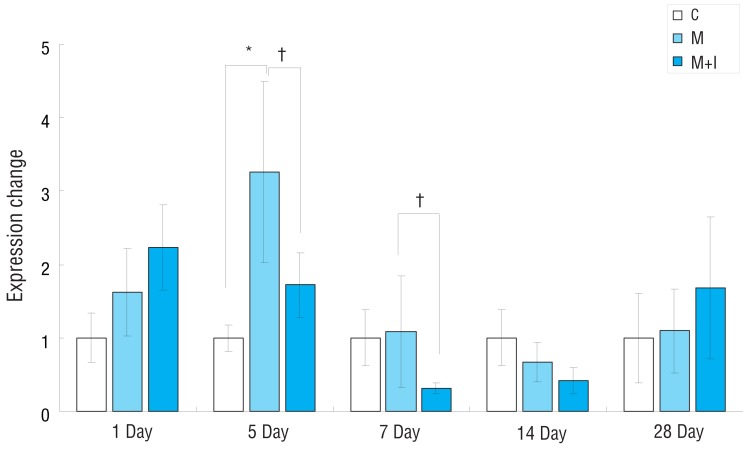
Gene expressions of TNF-α had significantly increased in the M group compared with the C group on day 5. Gene expressions of TNF-α had significantly decreased in the M+I group compared with the M group on days 5 and 7 (Fig. 2).
2) Gene expressions of ET-1
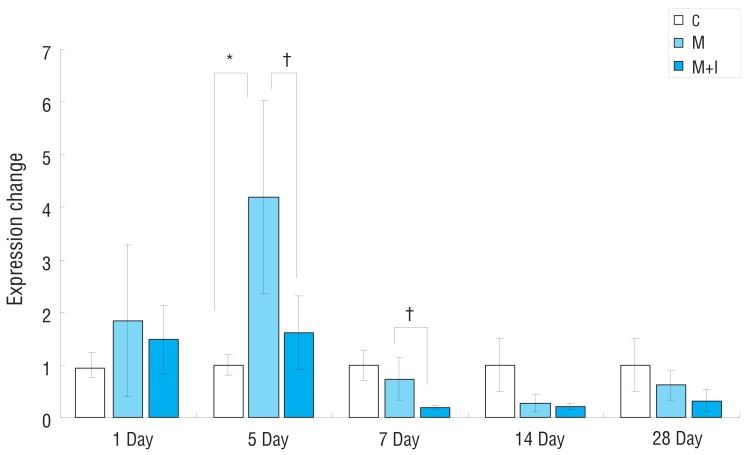
Gene expressions of ET-1 had significantly increased in the M group compared with the C group on day 5. Gene expressions of ET-1 in the M+I group was significantly lower compared with the M group on days 5 and 7 (Fig. 3).
3) Gene expressions of ERA
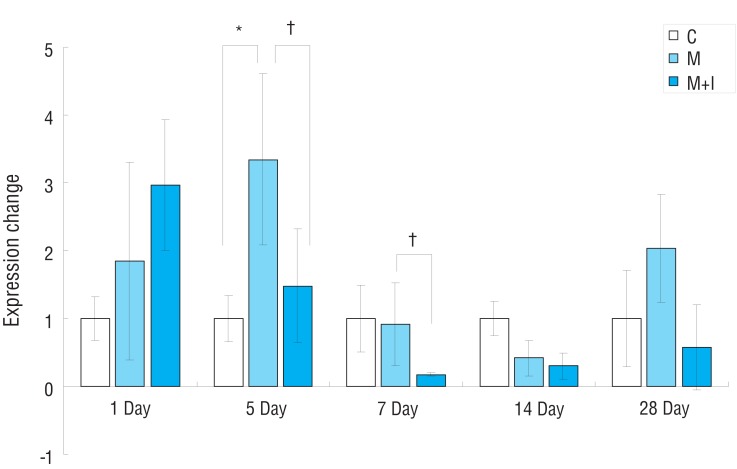
In the M group, gene expressions of ERA had significantly increased compared with the C group on day 5. Gene expressions of ERA had significantly decreased in the M+I group compared with the M group on days 5 and 7 (Fig. 4).
4) Gene expressions of eNOS
Gene expressions of eNOS had increased in the M group compared with the C group on days 7 and 28. Gene expressions of eNOS had significantly decreased in the M+I group compared than the M group on day 7 (Fig. 5).
5) Gene expressions of MMP2
Gene expressions of MMP2 had significantly increased in the M group compared with the C group on days 1, 5 and 28. Gene expressions of MMP2 had significantly decreased in the M+I group compared with the M group on day 5 (Fig. 6).
6) Gene expressions of TIMP
Gene expressions of TIMP had significantly increased in the M group compared with the control group on day 28. There were no significant changes between the M group and the M+I group (Fig. 7).
Discussion
In this study, pulmonary hypertension was induced by monocrotaline. In our previous studies have presented same result14-16). The evidence includes: a progressive increase in the ratio of RV to LV+S which was significantly noted and that the RVP had progressively increased in the M group compared with the C group. The pathologic findings were consistent with other results which showed a significant rise of pulmonary blood pressure and apparent RVH after monocrotaline injection9,17). The body weight of M and M+I group was lower than C group. Monocrotaline administration may lead to growth failure of rats in both M and M+I group. Therefore LV+S weight in M and M+I group was lower than C group. We thought RV/LV+S ratio comparison was more important to our experiment.
In this study, we described the effect of pathologic changes in infliximab on monocrotaline induced pulmonary hypertension rat models. Despite our expectations that infliximab would improve the ratio of RV/LV+S, there were no significant differences in RV and the ratio of RV/LV+S between the M group and the M+I group.
But, the mean RVP was decreased in the M+I group compared with the M group on days 14 and 28, but it was not statistically significant. The predominant changes in pulmonary vasculature included the development of medial wall thickening in the pulmonary arterioles and an increasing number of intra-acinar muscular pulmonary arteries in the M group. The changes of medial wall thickness were significantly attenuated in the M+I group on day 7 compared with the M+I group. The number of intra-acinar muscular arteries was significantly decreased in the M+I group on days 14 and 28. These vasculature changes may bring the changes of RVP, without anatomical changes. A longer follow up period would be needed to demonstrate the structural change of the RV after hemodynamic change.
We suggest an additional infliximab booster is needed on day 7 after an injection of monocrotaline and infliximab for better histological and hemodynamic results.
The administration of infliximab attenuated pulmonary arterial wall thickness on day 7 and reduced the number of arteries to the same level as the C group. This finding may represent neo-muscularization of non-muscular pulmonary arteries in the distal area to respiratory bronchioles in PAH. Schermuly et al.18) reported similar results that reduced medial thickening of the precapillary lung arteries under sildenafil and significant reduction of fully muscularized peripheral pulmonary arteries.
Our study showed infliximab reduced the number of pulmonary arteriole. Booth et al.19) reported that anti-TNF-α therapy resulted in clinical remission, reduced inflammation and improved endothelium-dependent vasomotor responses.
Infliximab decreased RVP and could be effective in pulmonary hypertension as a result of the prevention of pulmonary arterial wall thickening, vascular proliferation and remodeling. However, the changes of gene expression are obvious in our study. Therefore, we presume that histologic changes were followed after molecular changes.
Gene expressions of ET-1, ERA, NOS2, eNOS, MMP and were significantly increased in the M group in this study. We found that the proinflammatory mechanism is important to the development of pulmonary hypertension similar to other unrevealed heart disease such as congestive heart failure (CHF). In a murine model, Xiong et al.20) reported that infliximab treatment attenuated elastic fiber disruption, macrophage infiltration, and MMP2 and MMP9 expression in aortic tissue. They also suggested that TNF-α plays a central role in regulating matrix remodeling and inflammation in the aortic wall leading to abdominal aortic aneurysm. TNF receptor activation within the myocardium can induce LV myocardial remodeling. The molecular trigger for TNF-α mediated LV remodeling may exist through MMP induction21).
TNF-α is a potent cytokine that has been implicated in many autoimmune diseases and CHF21,22). The rationale for using TNF-α antagonists in systemic sclerosis associated with pulmonary hypertension arises from overproduction of this cytokine in the disease and contribution to lung interstitial and vascular damage23). Soluble TNF-α is generally regarded as an endogenous mediator of inflammation, modulating a wide spectrum of cellular responses including activation of genes involved in inflammatory and immunoregulatory responses, cell proliferation, antiviral responses, growth inhibition and cell death21). Infliximab, TNF-α antagonist induces caspase-dependent apoptosis of inflammatory cells and affects monocyte cytokine production. TNF-α antagonist also induces apoptosis of activated monocytes and T lymphocytes in Crohn's disease24) and other autoimmune disease such as rheumatic arthritis and sarcoidosis8,22,23).
In our present study, the gene expression of TNF-α had increased on day 5 in the M group and had been downregulated by infliximab on days 5 and 7.
TNF-α serves as a pivotal modulator of arteriogenesis as it is attenuated by treatment with TNF-α inhibitors. Grundmann et al.25) showed that infliximab inhibited arteriogenesis and lead to a decrease in mean collateral diameter, a proliferation of vascular smooth muscle cells and a reduction in leukocyte accumulation around collateral arteries.
The MMPs have been demonstrated to cause tissue remodeling in normal physiologic processes such as tissue morphogenesis, trophoblast migration and wound healing. Increased MMP expressions have been identified in the pathological process such as tumor angiogenesis and metastasis, rheumatoid arthritis, atheroma formation and plaque rupture26). Similar findings were examined on the gene expressions of MMP and TIMP. In our study, the MMP2 gene expression had increased, but significant downregulation occurred by infliximab. The gene expressions of TIMP showed no significant changes between the M group and the M+I group. Only significant increases of the M group compared with the C group appeared on day 28. Activation of latent MMPs is required for proteolytic activity. A loss of MMP inhibitory control through TIMP-1 gene deletion has been shown to cause LV dilatation in mice27). The results of this gene expression study imply that the morphological changes of pulmonary arterioles on monocrotaline-induced PAH result from changes in inhibition of these cytokine genes. The same result was shown in CHF. The plasma MMP levels had increased in CHF patients and they were related to temporal changes in TNF receptor activation21).
Vascular endothelium produces multiple vasoactive substances including NO, PG2, ET-1 and endothelium-dependent hyperpolarization factors28). ET-1 is the most potent vasoconstrictor and it is critical in the pathogenesis of vascular remodeling. An imbalance of dilators (NO and PG2) and constrictors (ET-1) may affect pulmonary vascular resistance in a variety of settings. Increased RVH and pulmonary arterial medial wall thickening was induced by an exposure to 60% O2 for 14 days and attenuated ETA/ETB receptor antagonist29). In our study, there was significant downregulation of ET-1 in the M+I group on day 5. The ERA gene expressions were also significantly decreased in the M+I group compared with the M group.
eNOS is involved in neoangiogenesis and increased in advanced pulmonary vascular lesions (Eisenmenger syndrome). NO is an important regulator of vascular tone in the pulmonary circulation. Hoehn et al.30) investigated NOS, inducible NOS (iNOS) and neuronal NOS in the lungs from biopsies of infants with pulmonary hypertension secondary to cardiac defects. They reported significant increases in eNOS and iNOS staining in pulmonary vascular endothelial cells and suggested upregulation of NOS3 and iNOS at an early stage of pulmonary hypertension. In our study, eNOS was not significantly increased except on day 28 in the M group. eNOS was decreased in the M+I group compared with the M group on day 28 but with no significant change. This finding could be due to the late gene expression of eNOS.
In our research, infliximab did not completely reverse monocrotaline-induced PAH. The effect was not sustained, and ultimately, rats treated with infliximab developed PAH. Our study revealed a weak inhibitory effect of infliximab on RVH and PAH. The effect of infliximab on attenuation of RVH and PAH seems to be weaker than that of bosentan21) and simvastatin31). But these studies had more frequent number of administration of medicine.
The dose of infliximab, used experimentally and clinically, was inconsistent among the various other investigators. The dosage and frequency of use of infliximab should be modified to prevent pulmonary hypertension in future research. Inflammatory cells and TNF-α are present in the remodeled arteries, and a study showed athymic rats model less developed monocrotaline induced PAH than euthymic rats model32).
Further broad studies are needed to investigate the changes of cytokine, apoptosis and vasoconstriction after administration of infliximab in a PAH rat model. The limitation of this study was the small number of subjects, which was due to mortality in rats after monocrotaline administration. More extension of the follow up period would be desirable to demonstrate improvement of RVH after decreased RVP.
In conclusion, in our research, the administration of TNF-α antagonist has a weak protective effect on PAH by inducing pulmonary vascular changes and having an influence on the gene expression in the inflammatory cytokines related to PAH.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


