




Graphical Abstract
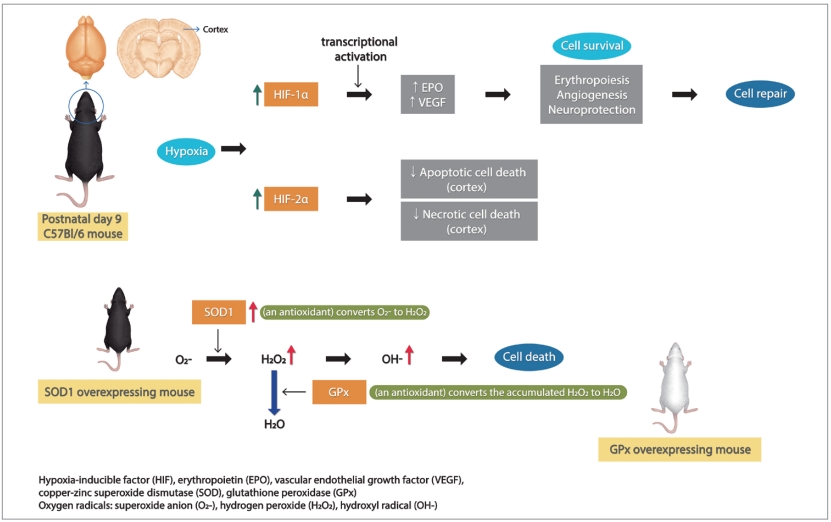
Mechanisms of cell survival mediated by HIF
Introduction
Neonatal hypoxic-ischemic encephalopathy causes death in twenty percent of affected infants, and 25% of survivors have permanent neurologic deficits such as developmental delay, cerebral palsy, cognitive dysfunction, hearing loss, and visual loss [1]. Ten percent of moderate encephalopathy infants die and 30% of the survivors are handicapped; 60% of severe encephalopathy infants die and most of the survivors have permanent disabilities [2]. Treatment of neonatal hypoxic-ischemic encephalopathy is limited to therapeutic hypothermia for those infants that qualify but does not offer complete protection. Several neuroprotective strategies have been suggested such as antioxidants or erythropoietin (EPO) [3].
We have previously reported that hypoxia-inducible factor (HIF)-1╬▒ deficient mice have increased brain injury after neonatal hypoxia-ischemia (HI) [4]. HIF-1╬▒ is a nuclear protein induced by hypoxia, which acts as a transcription factor crucial for activation of many protective hypoxia-inducible genes such as erythropoietin, glucose transporters, nducible nitric oxide synthas (NOS), and vascular endothelial growth factor (VEGF) [5,6]. As a result, HIF-1╬▒ promotes cell survival, and initiates neuroprotection, angiogenesis and subsequently, cell repair in neonatalHI [4,7].
We also have found that copper-zinc superoxide dismutase (SOD)1 overexpression is not beneficial to the neonatal mouse brain with HI injury [8], unlike the adult brain with similar injury, in which SOD1 overexpression was shown to be neuroprotective after cerebral ischemia [9]. SOD1 is an endogenous antioxidant, which converts superoxide anion (O2-) to hydrogen peroxide (H2O2) in neurons of the HI brain. Glutathione peroxidase (GPx) converts the accumulated H2O2 to H2O and O2. However, SOD1 overexpression causes high H2O2 accumulation after HI in the neonatal brain, but not the adult [10]. The accumulation of H2O2 and hydroxyl radical (OH-) causes nitric oxide radical production by NOS, lipid peroxidation, protein oxidation, DNA damage, and finally neuronal death via necrosis or apoptosis [11]. These data illustrate that neuroprotective mechanisms of the developing neonatal brain are different from mature brain. We explored whether the expression of HIF-1╬▒ and HIF-2╬▒ were altered in SOD1 overexpressing mouse brain compared to wild-type littermates to explain this increased injury in neonatal brain.
Methods
This animal study was approved by the University of California San Francisco (UCSF) institutional animal care and use committee in accordance with National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. C57Bl/6 mice and SOD1 overexpressing littermates were subjected to the Vannucci model of neonatal HI [12-14]. At postnatal day (P) 9, pups were anesthetized with isoflurane (3% isoflurane/balance oxygen), the left common carotid artery was dissected after a midline incision of the neck and the artery was ligated with electrical coagulation. Animals were allowed to recover for 1 hour with the dam and then exposed to 50 minutes of hypoxia in a humidified chamber at 36.5┬░C with 10% oxygen/balance nitrogen. Sham-operated control animals received isoflurane anesthesia and exposure of the left common carotid artery without ligation or hypoxia. Pups were sacrificed at designated times after injury (30 minutes, 4 hours, or 24 hours) by decapitation after intraperitoneal injection of Euthasol. Brains cortices were dissected on a cold pad, placed in labeled prechilled microfuge tubes, flash frozen in methylbutane on dry ice, then stored at -80┬░C until use. Animals used in each group were 10, 11, 15, 13, 7, 13, 10, and 13, in wild-type sham, SOD1 overexpressor sham, wild-type 30 minutes, SOD1 overexpressor 30 minutes, wild-type 4 hours, SOD1 overexpressor 4 hours, wild-type 24 hours, and SOD1 overexpressor 24 hours after HI, respectively.
Western blot analysis was performed on ipsilateral cortex from wild-type and SOD1 overexpressing mice to quantify HIF-1╬▒, HIF-2╬▒, spectrin 120 as a neuronal apoptosis marker, and spectrin 145/ 150 as a neuronal necrosis marker at 30 minutes, 4 hours, and 24 hours after HI injury.
Frozen cortices were homogenized and cytoplasmic and nuclear proteins were extracted with NE-PER (nuclear and cytoplasmic extraction reagents, Pierce, Rockford, IL, USA). Protein samples were prepared with 4X loading buffer and sample reducing agent (10X DTT) and 40-╬╝g cytoplasmic protein or 20-╬╝g nuclear protein underwent sodium dodecyl sulfate-polyacrylamide gel electrophoresis at 150 V for 90 minutes. Proteins were transferred to polyvinyl difluoride membranes (Bio-Rad, Hercules, CA, USA) at 35 V overnight at 4┬░C as described by Chang et al.15) The membranes were blocked in 5% nonfat dry milk in Tris-buffered saline with Tween for at least 1 hour at room temperature with gentle shaking, then incubated with rabbit HIF-1╬▒ polyclonal antibody (1:1500; Novus Biologicals, Littleton, CO, USA), rabbit HIF-2╬▒ polyclonal antibody (1:500; Novus Biologicals), and rabbit histone monoclonal antibody (1: 8000; Cell Signaling Technology, Boston, MA, USA) or mouse spectrin monoclonal antibody (1:2000; Millipore, Temecula, CA, USA) and mouse beta-actin monoclonal antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4┬░C. horseradish peroxidase-conjugated goat anti-rabbit antibodies (Santa Cruz Biotechnology) were used (1:2000 for HIF-1╬▒ and HIF-2╬▒, 1:4,000 for histone) as secondary antibodies. Bound antibodies were developed by enhanced chemiluminescence (ECL, Amersham Life Science, Arlington Heights, IL, USA) and exposed to film. Image J software (NIH, Bethesda, MD, USA) was used to measure the optical densities (OD) and areas of protein signal on after scanning.
Data were analyzed by nonparametric tests: the Mann-Whitney U test for 2 groups, and the analysis of variance with Kruskal-Wallis test for 3 or more groups. All data were analyzed using Prism 7 (GraphPad Software, La Jolla, CA, USA). Data are given as mean┬▒ standard error, and P values <0.05 were considered significant.
Results
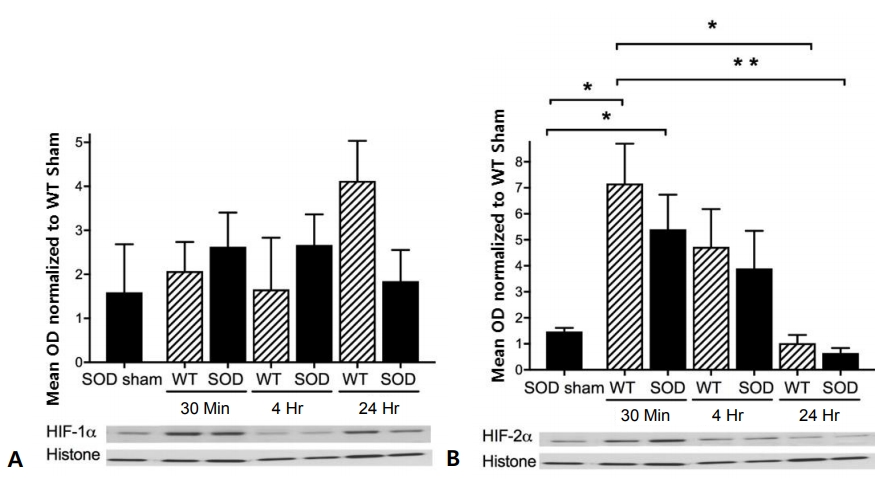
HIF-1╬▒ protein expression was not significantly altered in wild-type or SOD1 overexpressing mouse cortex after HI injury (Fig. 1A). However, HIF-2╬▒ protein expression increased in both wild-type and SOD1 overexpressing mouse cortex, reaching peak levels of 7-fold for wild-type and 5-fold for SOD1 overexpressors at 30 minutes after HI injury (Fig. 1B). The level of HIF-2╬▒ decreased at 4 hours and returned to baseline at 24 hours (Fig. 1B). There were no differences between SOD1 overexpressors and wild-type littermates by time point.
Spectrin 145/150 protein expression was not significantly altered in wild-type or SOD1 overexpressing mouse cortex after HI injury (Fig. 2A). However, spectrin 120 protein expression increased in both wild-type and SOD1 overexpressing mouse cortex, reaching peak levels of 3-fold for both wild-type and SOD1 overexpressors 4 hours after HI injury (Fig. 2B).
Discussion
The developing neonatal brain, with a high rate of oxygen consumption, high concentrations of unsaturated fatty acids and relative lack of antioxidants is extremely vulnerable to oxidative stress and exposure to exogenous free radicals and iron-mediated free radical injury [16]. Antioxidants protect the brain by attacking various injury cascades. These are reactive oxygen species scavengers (melatonin, SOD, catalase), inhibitors of lipid peroxidation, EPO (as a free radical reducer), inhibitors of NOS and anti-inflammatories [17]. The mechanism of neuroprotection by an iron chelator such as deferoxamine is that it activates and upregulates HIF-1╬▒ expression. The upregulated HIF-1╬▒ in turn activates various HIF-1╬▒ mediated genes, such as VEGF, which initiate a complicated neuroprotective cascade [18,19]. HIF-1╬▒, a subunit of HIF-1, is undetectable under normoxic conditions, which is induced by hypoxia and is promptly degraded under normoxia by the ubiquitin-proteasome pathway [20]. Like HIF-1╬▒, HIF-2╬▒ is induced by hypoxia. It is a less well-characterized factor, but has been shown to be expressed much more in astrocytes than neurons [21] and is the primary mediator of EPO expression induced by hypoxia, which is in turn protective to neurons [22], suggesting HIF-2╬▒ expression may play a bigger role in neuroprotection than HIF-1╬▒. The actions of these 2 HIF isoforms are cell-specific, regional and developmentally regulated, being complementary rather than redundant [23].
Previously, in P7 mouse cortex, we have seen increased HIF-1╬▒ immediately after hypoxia alone and 24 hours after HI [4]. Similarly, also in P7 mouse cortex, we saw increased HIF-1╬▒ immediately after hypoxia as well as at 4 hours and 24 hours after hypoxia [24]. However, HIF-1╬▒ increased only immediately after HI, not at 4 hours or 24 hours after HI [24]. On the contrary, HIF-1╬▒ decreased 4 hours after HI in P9 mouse cortex with no change in the hippocampus [25]. We also measured HIF-2╬▒ in the latter study. HIF-2╬▒ did not change after HI in cortex, but declined in hippocampus at 30 minutes, 4 hours, and 24 hours. These results corresponded with increased spectrin 145/150 at 4 hours and 24 hours in cortex, but not hippocampus, and with increased spectrin 120 at 24 hours in cortex and at 4 hours in hippocampus [25]. suggesting early and prolonged necrotic cell death with additional late stage apoptosis in the cortex, but early apoptotic cell death with a relative lack of necrotic cell death in the hippocampus. Thus, while we did not see significant changes in HIF-1╬▒ in the present study, we did see a strong increase in HIF-2╬▒ in both wild-type and SOD1 overexpressing cortex after HI. This may be due, in part, to the above-mentioned cell-specific, regional and complementary actions of the HIF isoforms. We measured spectrin here also in order to determine if mechanism of cell death corresponded to levels of HIF-1╬▒ or HIF-2╬▒. Since spectrin 145/150 was not elevated, but spectrin 120 was (at 4 hours), this may indicate that apoptosis has a greater role than necrosis in genesis of the HI injury in the critical phase several hours after the insult. The expression of HIF-2╬▒ may play a bigger role in neonatal HI injury than HIF-1╬▒. It is conceivable that HIF-2╬▒ stimulated production of factors (such as EPO) that protected neurons from necrotic injury while astrocytes (and perhaps neurons) underwent subsequent apoptotic death. While we did not analyze cell type, neurons and astrocytes may have different responses to injury that are reflected in gene expression at the time points studied.
The SOD1 overexpressing cortex behaved quite similarly to the wild-type cortex with regard to expression of HIF-1╬▒, HIF-2╬▒, spectrin 145/150, and spectrin 120 at the time points after HI analyzed here. Thus, alterations in expression of these factors do not explainthe increased injury we have previously seen in the SOD1 overexpressing brain compared to wild-type.
HIF-2╬▒ activates transcription of mitochondrial manganese SOD (SOD2) [26]. The early increase in HIF-2╬▒ seen in both wild-type and SOD1 overexpressing cortex may indicate additional SOD production; a factor that warrants further study.
The activation of HIF-1╬▒ and the consequence of the activation of its target genes are suggested to be necessary for protection in neonatal HI [4,11]. During neonatal brain injury, excitotoxicity (excessive activation of glutamate neurotransmission, oxidative stress, and inflammation) contributes to cell death either by apoptosis or necrosis. Apoptosis, or programmed cell death, seems to play a more important role than necrosis during HI injury [16].
The role of HIF-2╬▒ in neonatal HI brain is relatively unexplored. HIF-2╬▒ is activated by hypoxia stimulated mitogen-activated protein kinase (MAPK)/ERK signaling pathway, leading to the increased transcription of various proliferation factors. HIF-2╬▒ directly binds the MAPK3 promoter and activates its function through the interaction of the transcription factor and its regulated genes [27]. In cardiac myocytes in adult mice, myocyte-specific HIF-2╬▒ induced the epithelial growth factor amphiregulin (AREG), activating AREG signaling, and finally protecting the myocardium from myocardial ischemia-reperfusion injury [28]. In the carotid body, intermittent hypoxia leads to increased expression of HIF-1╬▒ and decreased levels of HIF-2╬▒ along with decreased SOD2 expression [29]. In mesenchymal stem cells, HIF-2╬▒ maintained significant long-term effects, and was more stable in hypoxic conditions than normoxic [27]. In the brain, HIF-2╬▒ is considered to be astrocyte specific, and astrocytes are important for the clearance of H2O2, resulting in neuroprotection. Wildtype brain with decreased expression of HIF-2╬▒ had more severe brain injury compared to GPx overexpressing brain with higher HIF-2╬▒. This may be a reflection of a neuroprotective effect of HIF-2╬▒ associated with SOD2 generated H2O2 clearing [25].
Increased HIF-1╬▒ after HI is reported in many cases [4,24,30], however, HIF-1╬▒ is not changed or rather decreased in some cases as previously mentioned [25]. The expressions of HIF-1╬▒ are different according to the time after HI, cell types (neurons vs. astrocyte), region (cortex vs. hippocampus), or developmental age (P7 vs. P9). So, these different experimental conditions partially account for these different results.
There are several limitations in this study. First, the grade of brain injury with histological analysis was not checked in the present study. Though we have shown injury severity with HI brain injury scores twice in the past [24], the experimental conditions such as developmental age (P7 vs. P9) were different from the previous study. It is not technically feasible to do histology and protein expression in the same sample. Also, spectrin is an assessment of injury which is not quite comparable to histology. Further work is needed to clarify the grade of brain injury. Second, SOD1 expression of SOD overexpressors was not checked. However, as these mice are well-characterized, and we have done SOD activity in the past [31], we didnŌĆÖt check SOD1 expression on the transgenics in the present study. Third, we could not find difference of protein expressions after HI between wild-type and SOD1 overexpressing mouse. Some of this might be due to small number of mice underwent this experiment.
In summary, we propose that both HIF-1╬▒ and HIF-2╬▒ may promote cell survival in neonatal HI, but in a cell-specific and regional fashion. We also suggest that early HIF-2╬▒ upregulation precedes apoptotic cell death and may limit necrotic cell death. Developmental age, degree and type of injury are all factors in the expression and action of HIF-1╬▒ and HIF-2╬▒. The influence of SOD was not clarified and remains an intriguing factor in neonatal HI. Further work is needed to clarify the roles of HIF-1╬▒ and HIF-2╬▒ in neonatal brain injury.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


