




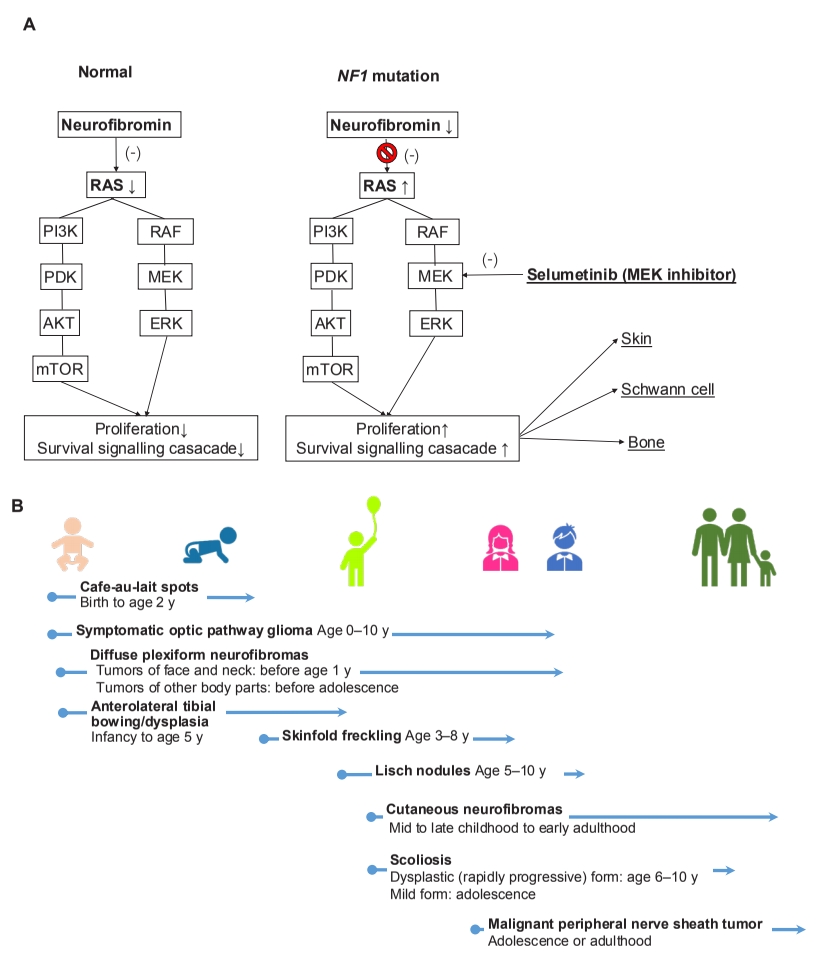
Neurofibromatosis 1 (NF1; MIM#162200) is an autosomal dominant genetic disease manifesting as multiorgan involvement from infancy to adulthood [1]. NF1 is caused by a pathogenic NF1 mutation with 100% penetrance that leads to the loss of neurofibromin function, which negatively regulates intracellular Ras–mitogen-activated protein kinases signaling (Fig. 1A) [2]. The manifestations mainly involve the skin and central or peripheral nervous system and are progressive with age [1,3]. Multiple café-au-lait spots, the first sign, presents by 1 year of age in almost all NF1 patients. In addition, approximately 50% of NF1 cases are sporadic, and only molecular testing is available to diagnose these patients since they do not meet National Institutes of Health (NIH) criteria by 1 year of age [1-6].
Although the diagnosis of NF1 is based on clinical findings according to the NIH criteria, the need for genetic testing is increasing for not only genetic counseling but also early diagnosis in very young patients who do not fulfill the NIH criteria [4,5]. Genetic testing also helps establish the genotype and phenotype correlation in NF1, and it also provides a plan for the surveillance of complications [6]. NF1 gene deletion or missense mutation of codons 844–848 is related to a severe phenotype including an increased risk of malignancy [6]. Single amino acid deletion at 2971 shows only café-au-lait spots and freckling, and missense mutations involving Arg1809 shows Noonan-like features including pulmonic stenosis [4,6]. Several conditions including Legius syndrome (MIM#611431), Silver–Russel syndrome (MIM# 180860), Proteus syndrome (MIM# 176920), Sotos syndrome (MIM#117550), neurofibromatosis type 2 (MIM#101000), constitutional mismatch repair deficiency syndrome (MIM# 276300), and Noonan syndrome (MIM#163950) could also present café-au-lait spots, a representative sign of NF1, and molecular testing helps differentiate NF1 from other genetic diseases [6,7].
In a recent issue of Clinical and Experimental Pediatrics, Kang et al. [8] reviewed the diagnosis and overall management of pediatric NF1 patients. The authors emphasized the importance of molecular genetics testing by pointing out the various systemic involvements and disease severities seen in NF1 patients. Further, they reported that it is difficult to diagnose NF1 early using clinical findings alone owing to age-dependent symptoms. In addition, after diagnosing NF1, the clinician can plan the timeline of systematic evaluation and introduce a multidisciplinary management approach including the new molecular target drug, selumetinib, which inhibits the MEK1 and MEK2 signaling hyperactivity [8]. The US National Cancer Institute reported that 71% (17 of 21) NF1 pediatric patients showed partial response to the MEK inhibitor selumetinib for reducing plexiform neurofibromas (PNs) in a clinical phase 1 trial; the phase 2 trial showed not only a similar partial response (70%) but also significant clinical benefits for pain (52%), motor dysfunction (66%), and dysfigurement (88%) among 50 pediatric NF1 patients with inoperative PNs [9,10]. Finally, in April 2020, the US Food and Drug Administration approved the use of selumetinib for pediatric NF1 patients (age>2 years) with symptomatic and inoperable PNs. With the advent of a new era of molecular target therapy in genetic disease pediatricians can diagnose patients early and promptly initiate proper aggressive management. Since the manifestation of NF1 starts even in infancy and progresses with age (Fig. 1B), timely education and regular surveillance is important to prevent complications. In the past, this genetic disease was subject to limited treatment options and prognosis, but it is now treatable with a well-targeted drug that offers meaningful results.
In conclusion, genetic testing should be considered for diagnosing NF1 in young children showing only multiple café-au-lait spots. After the diagnosis is established, annual or more frequent follow-up of all NF1 patients is recommended to prevent complications.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


