




Introduction
Neurofibromatosis type 1 (NF1; OMIM#162200) is a prevalent genetic disease that affects 1 in 3,000 individuals. NF1 is transmitted in an autosomal dominant manner and characterized by multiple cutaneous café-au-lait spots and neurofibromas. Variable degree of neurological, skeletal, and neoplastic manifestations are also noted. Affected patients are at risk of developing tumors of the central and peripheral nervous systems [1,2].
The phenotypes of several genetic diseases, especially those with autosomal dominant inheritance, are widely heterogeneous because of the differences in penetrance and expressivity among patients. NF1 is a genetic disorder with high penetrance, and almost all affected persons have café-au-lait spots; however, the extent of cutaneous lesions is highly variable among patients, e.g., its range of expressivity is wide [3].
Such phenotypic heterogeneity renders NF1 difficult to diagnose. In such situations, genetic tests provide important information about its diagnosis and prognosis as well as future reproductive options for family members. Age-appropriate surveillance and management are important to improving the quality of life of affected patients.
This review suggests several points from the clinical and molecular genetic perspectives to help general pediatricians provide adequate clinical care and genetic counseling to patients with NF1 and their families.
Diagnosis of neurofibromatosis type I
A diagnosis of NF1 is based on National Institutes of Health (NIH) criteria [4]. The important clinical features of its diagnosis include the size and number of café-au-lait spots and freckling, cutaneous neurofibromas, Lisch nodules in the eyes, and family history. However, some patients do not meet the NIH criteria for diagnosis; for example, some pediatric patients have only a few café-au-lait spots of variable size. By the age of 1 year, only about 50% of infants without a family history of NF1 will meet these criteria [5].
Because the frequency of clinical NF1 features increases with age, a clinical diagnosis cannot always be determined in pediatric patients. Overt NF1 manifestations such as optic pathway glioma and skeletal dysplasias, such as sphenoid dysplasia or tibial pseudarthrosis, can help confirm a diagnosis in such patients. Other clinical features suggestive of NF1 include macrocephaly and areas of focal abnormal signal intensity (FASI) in the brain, which have been detected in 59.5% and 87.0% of Korean pediatric patients aged <8 years with NF1, respectively [3].
Genetic tests can help confirm the diagnosis of NF1. Only the NF1 gene, which contains 57 constitutive exons and at least 3 alternatively spliced exons, is responsible for NF1. It is a large gene with highly homologous NF1 pseudogenes that interfere with genetic tests. The direct sequencing of genomic DNA alone has a detection rate of ~60% in patients with NF1 [6,7]. Therefore, multistep analyses of genomic and complementary DNA and changes in exon copy numbers have been recommended as standard tests, and their mutation detection rate is 95% among patients with NF1 [8-10]. However, these methods are labor-intensive; thus, we developed long-range polymerase chain reaction (PCR) and multiplex ligation-dependent probe amplification analyses of genomic DNA and identified NF1 mutations in 87.1% of 389 Korean families [3].
Still, a subset of patients with NF1 has no known NF1 mutations but might have germline mutations in the regulatory or splicing regions of the NF1 gene or somatic mosaic NF1 genetic mutations. Conventional PCR and sequencing techniques might overlook these mutations because their mutation burden is low; moreover, these techniques are mostly qualitative and not quantitative.
Another point to consider in diagnosis workups is that patients could be affected by other genetic diseases such as Legius syndrome (OMIM#611431), a constitutional mismatch repair deficiency (OMIM#276300), NF II (OMIM#101000), Noonan syndrome with multiple lentigines (OMIM#151100), multiple café-au-lait spots (OMIM#114030), and partial unilateral lentinogenesis. Some patients with these disorders might meet the diagnostic criteria of NF1. However, Lisch nodules and cutaneous or internal neurofibromas are not associated with any of these conditions. Nonetheless, NF1 is a lifelong evolving disease, and pediatric patients might not manifest its full spectrum. Therefore, a molecular differential diagnosis can help. The use of magnetic resonance imaging to identify brain areas with FASI also helps in the diagnosis.
Approximately 8% of pediatric patients with café-au-lait spots but no other clinical features of NF1 might have Legius syndrome, which is caused by a heterozygous mutation in the SPRED1 gene that enhances Ras inactivation by interacting with neurofibromin and translocates neurofibromin from the cytosol to membrane-anchored Ras [11]. Therefore, genetic testing for the SPRED1 gene can be considered for patients with café-au-lait spots but no NF1 gene mutations. A small subset of patients has café-au-lait spots but no mutations in the NF1 gene or any other genes responsible for the related genetic diseases. Multiple café-au-lait spots (OMIM#114030) can be inherited in an autosomal dominant manner. Whether this benign condition is a separate genetic disease remains to be determined, and a genetic cause has not been identified.
Value of genetic diagnosis
NF1 can be diagnosed based on the NIH criteria without genetic testing. Thus, patients or their family members might question physicians about the value of genetic testing for NF1.
Counseling is critically important during the process of genetic testing for NF1. Pretest counseling should include informing patients and/or their parents that mutation detection rates differ among test methods and that negative results do not necessarily exclude NF1. The identification of a pathogenic mutation provides important information than can predict the natural course of the disease. For example, a whole NF1 gene deletion or a haploinsufficient NF1 gene is associated with the early manifestation of neurofibromas, more frequent and severe intellectual disabilities, and dysmorphic facial features [11,12], and a 3-bp inframe deletion in exon 17 (c.2970-2972delAAT) is associated with a milder NF1 phenotype without cutaneous neurofibromas [13]. Missense variants at Arg1809 are associated with multiple café-au-lait spots and Noonan syndrome–like features [14,15]. Moreover, plexiform neurofibromas (PN), symptomatic spinal neurofibromas, optic pathway gliomas, and skeletal abnormalities are more frequently seen in patients with a missense mutation at codons 844–848 [16]. However, besides these correlations between specific genotypes and phenotypes, the overall genotype-phenotype correlations in NF1 have remained elusive. We recently classified overall correlations in a large cohort of Korean patients with NF1 [3]. Severe NF1 phenotypes were classified as being in a distinct or “NF1-plus” (NF1+) subgroup. The features of this group comprise manifestations considered clinically severe and requiring medical attention. Such manifestations include widespread diffuse cutaneous neurofibromas, learning disabilities, autism, seizures, cardiac abnormalities, hearing defects, optic pathway gliomas, severe PN (>3 cm in diameter) accompanied by disfigurement, pain, bony destruction, or located in the para-aortic area, brain tumors, nerve root tumors, malignant peripheral nerve sheath tumors, moyamoya disease, and bony dysplasia.
This subclassification led to the detection of a higher prevalence of NF1+ in patients with disruptive NF1 mutations including NF1 haploinsufficiency; frameshift, nonsense, and splicing mutations; and a lower prevalence in patients with missense/inframe NF1 mutations (64.3% in large deletions, 59.6% in truncating/splicing mutations, and 36.6% in missense/inframe mutations, P=0.001) [3]. These findings provided important information in terms of genetic counseling for families as well as patients with NF1. As NF1 is mostly diagnosed in children, parents are concerned about their child’s long-term prognosis. Because NF1 is a lifelong evolving disease, the natural outcomes of affected patients are difficult to predict based on the clinical features evaluated at the time of diagnosis. In these settings, knowledge of the genotype responsible for NF1 in individual patients might help predict the severity of their natural clinical course. However, although NF1 is a highly penetrant autosomal dominant disease, the phenotypic expressivity is wide among patients and even within family members with the same genotype.
Another benefit of genetic diagnosis is the availability of reproductive options for the affected patients or their family members. Approximately 50% of patients with NF1 have no known family history of NF1 [5], and 70% of NF1 cases in Korean patients are sporadic [3]. Prenatal genetic tests can be considered for the parents of a child with sporadic NF1 who wish to have more children in the future since germline mosaicism remains possible in either parent. Prenatal genetic tests or a preimplantation genetic diagnosis can be considered if an adult patient with NF1 plans a pregnancy because the risk of NF1 transmission from either parent to the baby is 50%. Families with a confirmed pathogenic NF1 mutation can choose to undergo prenatal tests and preimplantation genetic diagnoses. Prenatal tests to be considered are chorionic villi sampling and amniocentesis or cord blood sampling during gestational weeks 10–13, 15–20, and 20–24, respectively. A preimplantation diagnosis can be achieved by genetic tests to identify a pathogenic NF1 mutation of concern in one or more cells removed from early embryos conceived by in vitro fertilization, followed by the transfer of embryo(s) without the mutation to the uterus.
Age-appropriate surveillance
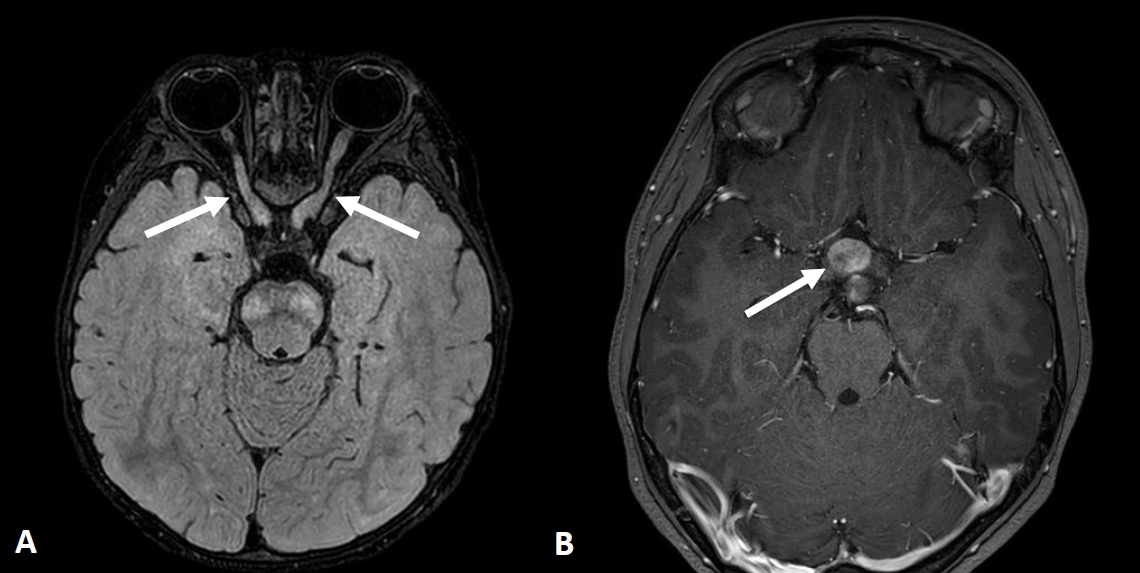
Because of the wide phenotypic NF1 heterogeneity and the fact that the NF1 spectrum evolves with age, the surveillance of clinical features must be age-appropriate. The spectrum of NF1 includes cutaneous, ocular, neurological, musculoskeletal, vascular and cardiac manifestations, and tumors (Table 1) [1,2,17-26]. Cutaneous symptoms including café-au-lait spots are common in pediatric patients, whereas cutaneous neurofibromas are not. Lisch nodules are melanocytic iris hamartomas, which are detectable in only 50% of pediatric patients and in ~75% of mid-teen patients [22]. Optic pathway gliomas are the most important ocular findings in pediatric patients (Fig. 1) because although most are asymptomatic, their progression can lead to a loss of visual acuity, proptosis, and strabismus.
Gross motor development is frequently delayed, and learning disabilities, intellectual deficits, and autism spectrum disorder might be encountered. Although seizures are infrequently encountered, the rates are higher in patients with NF1 than in the general population. Most individuals with NF1 have normal intelligence. However, learning deficits or behavioral problems and attention deficits are evident in 50%–80% of affected patients [27,28].
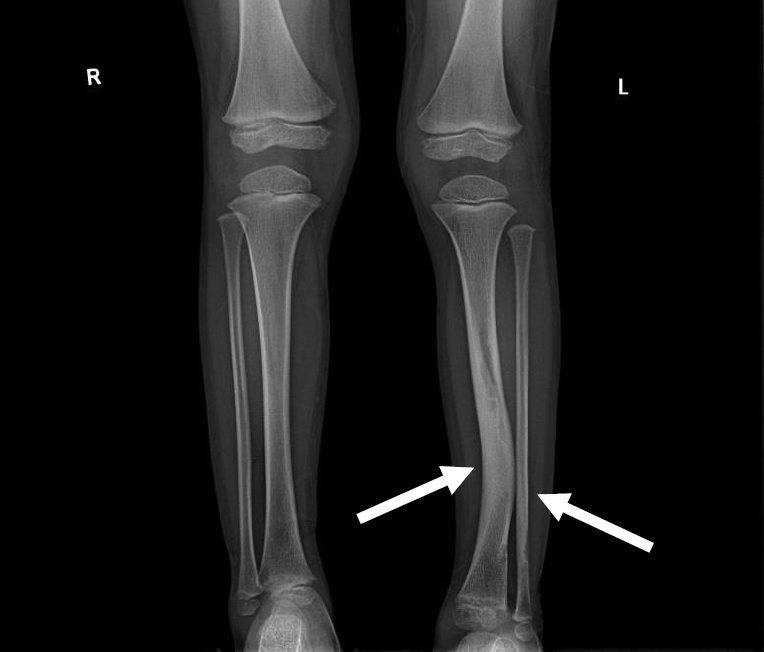
Osteopenia with vitamin D deficiency is another frequent manifestation, and skeletal dysplasia usually affects the sphenoid bones, lower legs, and vertebrae (Fig. 2). Scoliosis is also more prevalent in NF1 than in the general population, and stenotic or ectatic vascular abnormalities can also emerge. Moyamoya disease is 3-fold more prevalent in children with NF1 than in the general population [25]. In addition, renal artery stenosis and arterial aneurysms can occur. The pathogenesis of NF1-related vasculopathy is poorly understood; however, impaired neurofibromin expression in vascular endothelial cells is likely to result in abnormal vascular proliferation and growth [29].
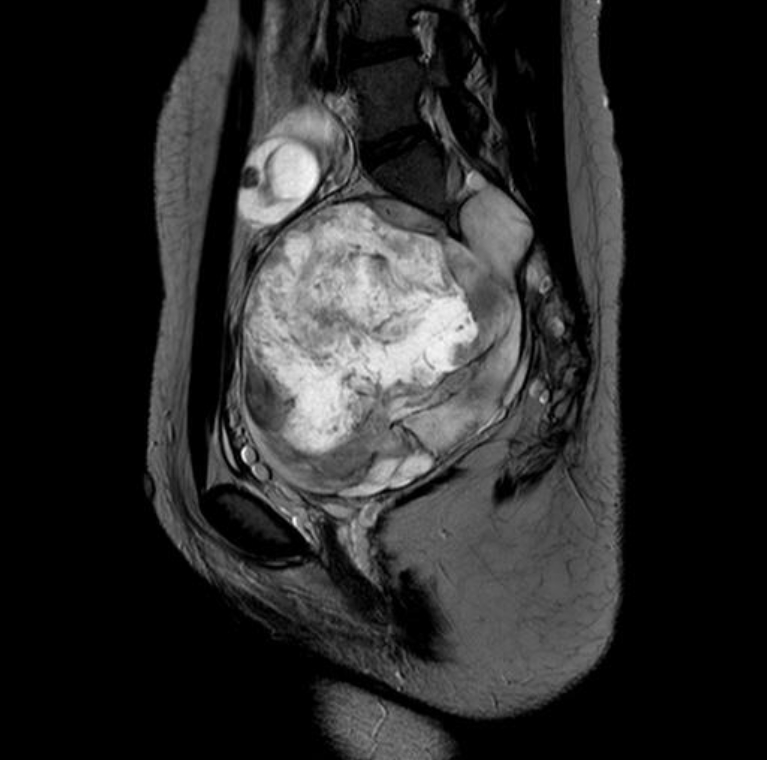
Tumors are the most critical complications of NF1. Cutaneous neurofibromas are benign, and their surgical removal can be recommended for selected patients. Patients with NF1 are at risk of developing tumors of the central and peripheral nervous systems, including PN (20%–30%), optic gliomas (~15%), pheochromocytomas (1%), and malignant peripheral nerve sheath tumors (5%) (Fig. 3) [1,2]. PN are benign nerve sheath tumors that result in devastating complications of NF1. They are thought to be congenital but might not be diagnosed until later in life with consistent growth. PN grow along nerves and involve multiple nerve branches and can cause significant morbidity owing to compressed vital structures, pain, disfigurement, and the risk of malignant transformation (Fig. 4). Surgery has been suggested as the only standard treatment for PN. However, up to 44% of tumors progress after the first surgery, especially in patients aged <10 years with head and neck tumors that are not completely resectable [21,23].
Management and selumetinib
The treatment of NF1 is mostly supportive and conservative. However, patients with clinical features of severe phenotypes (NF1+) [3] require referral to an appropriate center that can provide genetic analysis, evaluate multiorgan involvement, and surgical treatment. Cutaneous neurofibromas can be surgically removed if they are continuously growing, causing pain or disfigurement, or patients elect to have them removed.
Regular neuropsychological assessment is needed. Whole-body magnetic resonance imaging is an efficient method of detecting brain, optic nerve, and vascular abnormalities as well as internal neurofibromas and other tumors.
Regular ophthalmological assessment is also recommended to survey the functional abnormalities associated with optic pathway glioma. Progressive optic pathway gliomas with visual impairment require chemotherapy, but the outcomes are controversial [18]. Vascular abnormalities should be treated according to the neurological or neurosurgical assessment findings.
Surgical treatment should be considered for symptomatic moyamoya disease associated with NF1 and severe scoliosis, but the outcomes of surgery for tibial pseudarthrosis remain unsatisfactory and the procedure is challenging [30].
PN is a devastating complication of NF1. Progressively growing PN must be removed because of its potential for malignant transformation. However, complete resection is impossible in a substantial number of patients [21,23]. Much effort has been directed to clinical trials with targeted agents [31] such as tipifarnib [32], pirfenidone [33], sirolimus [34], pegylated interferon α-2b [35], and imatinib [36] (Table 2). Among these trials, a decrease in the tumor volume ≥20% from baseline was identified in only 4 of 83 patients (5%) in the pegylated interferon alfa-2b trial [35] and in only 6 of 36 (17%) in the imatinib trial [36].
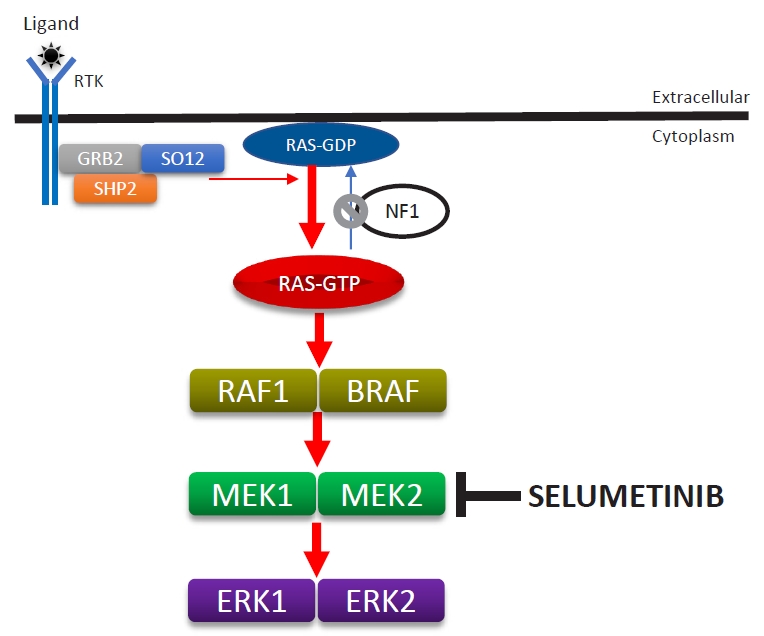
NF1 is caused by a germline loss of function of the NF1 gene at 17q11.2. The NF1 gene encodes neurofibromin, a tumor suppressor that regulates Ras activity via the hydrolysis of RAS-GTP to RAS-GDP [37]. Loss of NF1 heterozygosity is associated with the development of PN in neoplastic Schwann cells [23,38]. Therefore, loss of neurofibromin is associated with elevated activated Ras levels. Activated RAS increases the activities of RAF, MEK, and ERK, which comprise an important signaling pathway for increased cell growth, proliferation, and differentiation (Fig. 5).
Selumetinib (AZD6244; AstraZeneca plc., Cambridge, UK), an oral selective MEK inhibitor, decreases PN size without serious adverse reactions in pediatric patients [39]. A phase II study of selumetinib (25 mg/m2) with a median of 30 cycles (28-day) showed tumor volume decreases from baseline of ≥20% in 71% of pediatric patients without disease progression as well as decreases from a baseline neurofibroma volume in 12 of 18 mice (67%). Disease progression (≥20% increase in tumor volume from baseline) has not yet been found. Asian patients might have had higher exposure to selumetinib than other populations, and a lower dosage might be safer for Asians than for other populations. However, only one study has investigated selumetinib pharmacokinetics in healthy adult Asians [40]. One ongoing clinical study is investigating the clinical safety, pharmacokinetic properties, and effects of selumetinib in Korean patients with NF1 and inoperable PN (https://cris.nih.go.kr/cris/en/search/search_result_st01.jsp?seq=13575; KCT0003700). The U.S. Food and Drug Administration approved selumetinib (Koselugo) for pediatric patients with symptomatic inoperable PN in April 2020, leading to the dawn of a new era in the treatment of NF1. Notably, selumetinib also exerts clinically beneficial effects on pediatric low-grade glioma [41].
In conclusion, multisystemic and lifelong surveillance is required for NF1, which is one of the most common genetic diseases. The introduction of new therapeutic agents will improve the quality of life and survival rates of affected patients, provoke further investigations into the usefulness of selumetinib for conditions other than PN, and aid in the development of new therapeutic agents.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


