




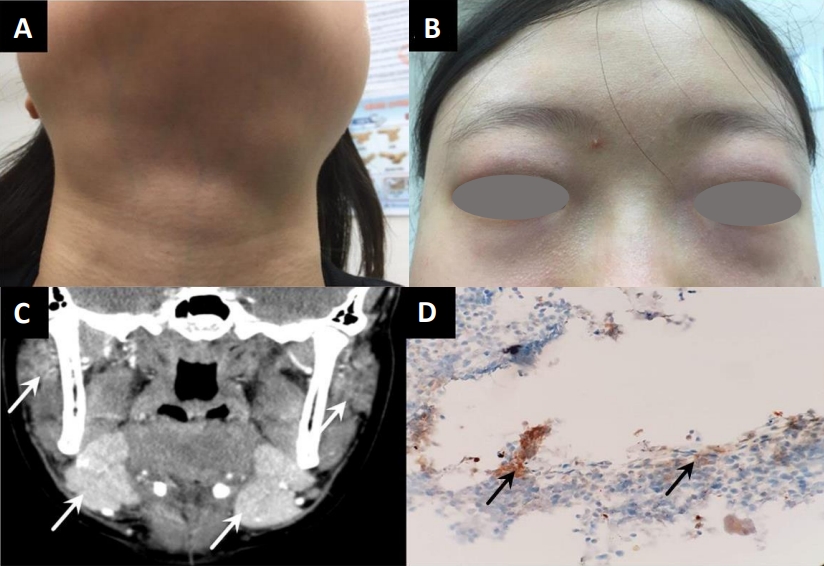
A 15-year-old female was evaluated for a persistent mass on the neck for 9 months. The patient presented with xerostomia and a dry cough. She denied constitutional symptoms including, fever, hyperhidrosis, weight loss, and abdominal pain. Physical examination confirmed nontender swellings of both parotid and submandibular glands (Fig. 1A). Both eyelids showed erythematous swelling with enlarged lacrimal glands (Fig. 1B). Bean-sized cervical lymph nodes were palpable on the left side. Breath sounds did not decrease and had no crackles or friction rub. The liver and spleen were not palpable below the costal margin. Furthermore, the patient had no abdominal tenderness. The laboratory findings are shown in Table 1. Plain chest radiography showed a small amount of pleural effusion in the left lower lung field. A computed tomography (CT) scan of the neck identified enlargement of the submandibular, parotid, and sublingual glands with lymphadenopathy (Fig. 1C). Submandibular gland biopsy revealed chronic inflammation, lymphoplasmacytic infiltration, slender fibrotic tissue, and more than 40 IgG4-positive cells per high-power field without obliterative phlebitis (Fig. 1D).
Based on the history and clinical findings, which of the following is the most likely diagnosis?
a. Sjögren syndrome
b. IgG4-RD
c. Systemic lupus erythematosus (SLE)
d. Lymphoma
Characteristics of patients with IgG4-RD
IgG4-RD is a recently recognized systemic immune-mediated fibroinflammatory condition caused by tumefactive lesions and infiltration of IgG4-bearing plasma cells that can affect several organs such as the salivary and lacrimal glands, pancreas, aorta, retroperitoneum, lung, and lymph nodes [1]. Clinical features such as enlargement of the salivary and lacrimal glands, as observed in our patient, would be previously considered Mikulicz disease. However, according to the recent definition of IgG4-RD, this condition is now classified as a subcategory of IgG4-related sialadenitis [2]. The most common form of IgG4-RD is type 1 autoimmune pancreatitis, but there are many other conditions that are considered manifestations of this disease, such as Mikulicz disease, lymphadenopathy, dacryoadenitis, chronic thyroiditis, interstitial pneumonia, sclerosing cholangitis, interstitial nephritis, aortitis, retroperitoneal fibrosis, and prostatitis [3].
Although the epidemiology of the disease is insufficiently described due to the lack of medical reports and broad array of symptoms, some demographic peculiarities have been found to stand out. Most patients are men over 50 years of age, which represents a discrepancy with other autoimmune diseases such as Sjögren syndrome and SLE.
Diagnosis of IgG4-RD is often delayed or unrecognized because of unawareness. In children, IgG4-RD is even more uncommon and will subsequently lead to significantly delayed diagnosis or unrecognized disease. Therefore, knowledge and awareness of this disease are essential to prevent missing the diagnosis and subsequent delay of treatment, especially in children.
Biological mechanism underlying the elevated IgG4 levels in IgG4-RD
Although the pathogenesis of IgG4-RD has not been completely elucidated, it is known that cytokines such as interleukin (IL)-4, IL-5, and IL-13 produced by type 2 helper (Th2) T cells and regulatory T cells are involved in the elevation of IgG4 levels [1]. The precise role of IgG4 in the pathogenesis of IgG4-RD remains unknown. Among the four subclasses of IgG, the least abundant is IgG4, ranging from 0.35 to 0.51 mg/mL [4]. The interactions of IgG4 with the Fc receptor and C1 are weaker than those of the other IgG subclasses [4]. Thus, IgG4 is less capable than the other subtypes in triggering immune responses such as phagocytic activation, antibody-dependent cellular cytotoxicity, and tissue damage caused by complement activation [5]. For these reasons, IgG4 is now thought to play a role in alleviating, rather than causing or aggravating the immune response. An emerging consensus is that elevated IgG4 levels in IgG4-RD may result from a down-regulatory response to primary processes such as Th2 cell activation [6].
Phenotypes of IgG4-RD
Unfortunately, the heterogeneity and multiorgan condition of these symptoms do not permit a clear and standardized identification, causing many delays and difficulties in the diagnosis. An attempt to identify distinct phenotypic groups is required to reduce diagnostic delays. Recently, a large cohort study was conducted in Japan [7], which showed that clinical manifestations of IgG4-RD could be divided into 4 phenotypes depending on organ involvement. The 4 phenotypes were as follows: group 1, pancreato-hepato-biliary disease; group 2, retroperitoneal fibrosis and/or aortitis; group 3, head and neck-limited disease; and group 4, classic Mikulicz syndrome with systemic involvement, accounting for 31%, 24%, 24%, and 22% of the IgG4-RD cases, respectively. It was also found that Asians and females are particularly predisposed to developing the head-and-neck-limited disease. In our study, we considered our patient to be in the ‘group 3’ category. It should be noted that a more accurate diagnosis of IgG4-RD could be achieved through the classification of patient-specific phenotypes. In the future, it will be necessary to elucidate the etiology of IgG4-RD, subdivide each phenotype, and determine appropriate treatment and prognosis.
Patient outcome
The patient was prescribed prednisolone 50 mg/day (1 mg/kg) for 1 month, followed by 35 mg/day for seven months, and 10 mg/day thereafter. One month after starting treatment, the serum IgG4 concentration decreased to 356 mg/dL, and repeat plain chest radiography showed improvement of pleural effusion. At the 3-month follow-up, the sizes of both the salivary and lacrimal glands were significantly reduced. The erythematous swelling around both eyelids and dry mouth also improved. The patient had no recurrence of symptoms associated with IgG4-RD at the most recent 18-month follow-up.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


